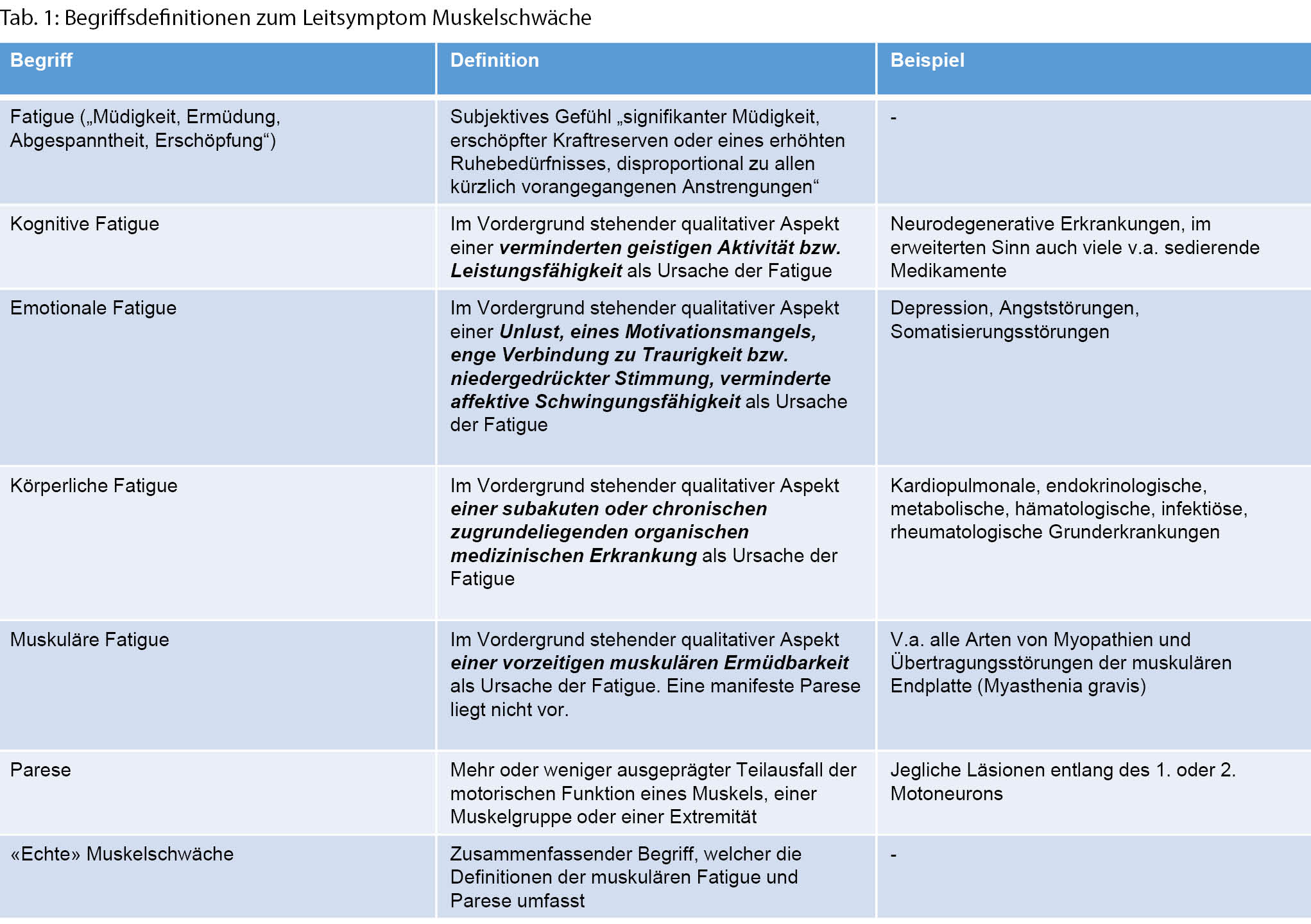
Muskelschwäche ist ein häufiges Symptom, das zur Vorstellung in einer allgemeinen Praxis führt. Die diagnostische Aufarbeitung beginnt mit der Unterscheidung einer «echten» Muskelschwäche von einer Fatigue. Die Lokalisationsverteilung, zeitliche Entwicklung und Schwere der Muskelschwäche können unter Beachtung von Begleitsymptomen und -erkrankungen, der Medikamenten- und Familienanamnese helfen, eine erste ätiologische Einordnung durchzuführen. Diese umfasst genetische, inflammatorische, infektiöse, neoplastische, toxische und metabolische/endokrine Ursachen als Hauptkategorien. Weitergehende Untersuchungen mit gezielten laborchemischen Abklärungen, ENMG, MRI, Muskelbiopsie und genetischer Abklärung können helfen, das weite Feld möglicher Differenzialdiagnosen genauer abzugrenzen. Insbesondere durch Fortschritte in der genetischen Abklärung und gezieltere immunmodulierende Therapie hat sich das Spektrum behandelbarer Erkrankungen, die mit einer Muskelschwäche einhergehen, in den vergangenen Jahren deutlich erweitert. .
Schlüsselwörter: Muskelschwäche, Fatigue, Myopathie, Parese
Einführung
Die Evaluation einer Person mit Muskelschwäche sollte im Allgemeinen drei Punkte umfassen [1]:
◆ Die Unterscheidung einer «echten» Muskelschwäche (Parese oder muskuläre Fatigue) von einer allgemeinen Mattigkeit/Müdigkeit (Fatigue mit emotionalen, kognitiven oder körperlichen Subtypen, s.a. Definitionen in Tab. 1) oder Bewegungseinschränkung, die nicht auf eine Reduktion der Muskelkraft zurückzuführen ist.
◆ Die Lokalisation der Muskelschwäche im neuromuskulären System («Mustererkennung» der Verteilung der Parese oder muskulären Fatigue).
◆ Die Feststellung der Ursache der Muskelschwäche (ätiologische Einordnung).
Unterscheidung einer «echten» Muskelschwäche von Fatigue
Ein häufiges Problem in der Praxis ist, dass eine Patientin/ ein Patient über «Muskelschwäche» klagt, ohne dass sich eine zu objektivierende Muskelschwäche (d. h. muskuläre Parese im eigentlichen Sinn oder muskuläre Fatigue) findet. Eine gezielte Anamnese und körperliche/neurologische Untersuchung können helfen, eine «echte» Muskelschwäche von einer erhöhten körperlichen oder kognitiven Ermüdbarkeit (Fatigue) oder mechanischen Bewegungseinschränkung, beispielsweise durch primär im Vordergrund stehende Schmerzen oder orthopädische/rheumatologische Beschwerden, abzugrenzen.
Anamnese
Diverse systemische Erkrankungen führen zu einer reduzierten Belastungsfähigkeit, die subjektiv als Schwäche bei bestimmten Aufgaben empfunden wird. Hierzu gehören u.a. kardiopulmonale Erkrankungen, rheumatologische/ orthopädische Beschwerden, eine Anämie, eine Kachexie durch maligne Erkrankungen, chronische infektiöse oder inflammatorische Erkrankungen (körperliche Fatigue) oder auch eine Depression mit Adynamie (emotionale Fatigue). Personen mit diesen Beschwerdebildern sind funktionell eingeschränkt, haben aber keine eigentliche Muskelschwäche im Sinne einer muskulären Parese.
Hauptbeschwerde bei Patientinnen und Patienten mit erhöhter körperlicher Ermüdbarkeit (Fatigue) ist ein (oft generalisiertes) körperliches Schwächegefühl und fehlende körperliche Belastungsfähigkeit, während Kranke mit Muskelschwäche typischerweise über aufgabenspezifische Einschränkungen (Einschränkungen beim Haarekämmen oder Treppensteigen) und/oder ein Gefühl von Schwere oder Steifigkeit in den Extremitäten klagen. Im Vordergrund stehende Muskelschmerzen sind bei neuromuskulären Erkrankungen relativ selten, bei Personen mit rheumatologischen Beschwerdebildern und Fibromyalgie dagegen häufig [5]. So steht beispielsweise bei einer Polymyalgia rheumatica eine schmerzhafte Bewegungseinschränkung und eine Steifheit im Vordergrund, während sich in der Untersuchung keine Einschränkung der Kraftgrade findet.
Untersuchung
Die Untersuchung ergänzt die in der Anamnese erhobenen Befunde durch die gezielte Suche nach Begleitzeichen, die eine subjektiv wahrgenommene Muskelermüdung von einer objektivierbaren Muskelschwäche abgrenzen können. Zwei Bemerkungen können hierbei hilfreich sein:
◆ Trotz einer fortgeschrittenen Muskelatrophie ist bei Patientinnen und Patienten mit Kachexie die Muskelkraft im Verhältnis zur Muskelmasse im Allgemeinen nicht eingeschränkt.
◆ Eine vermehrte Druckempfindlichkeit der Muskulatur ist in der Regel nicht mit einer «echten» Muskelschwäche assoziiert. Seltene Ausnahmen bilden hier infektiöse Ursachen (Trichinellose, virale Myositis), bestimmte toxische Myopathien, eine Myopathie hervorgerufen durch Störungen der Schilddrüsenfunktion und hereditäre metabolische Myopathien.
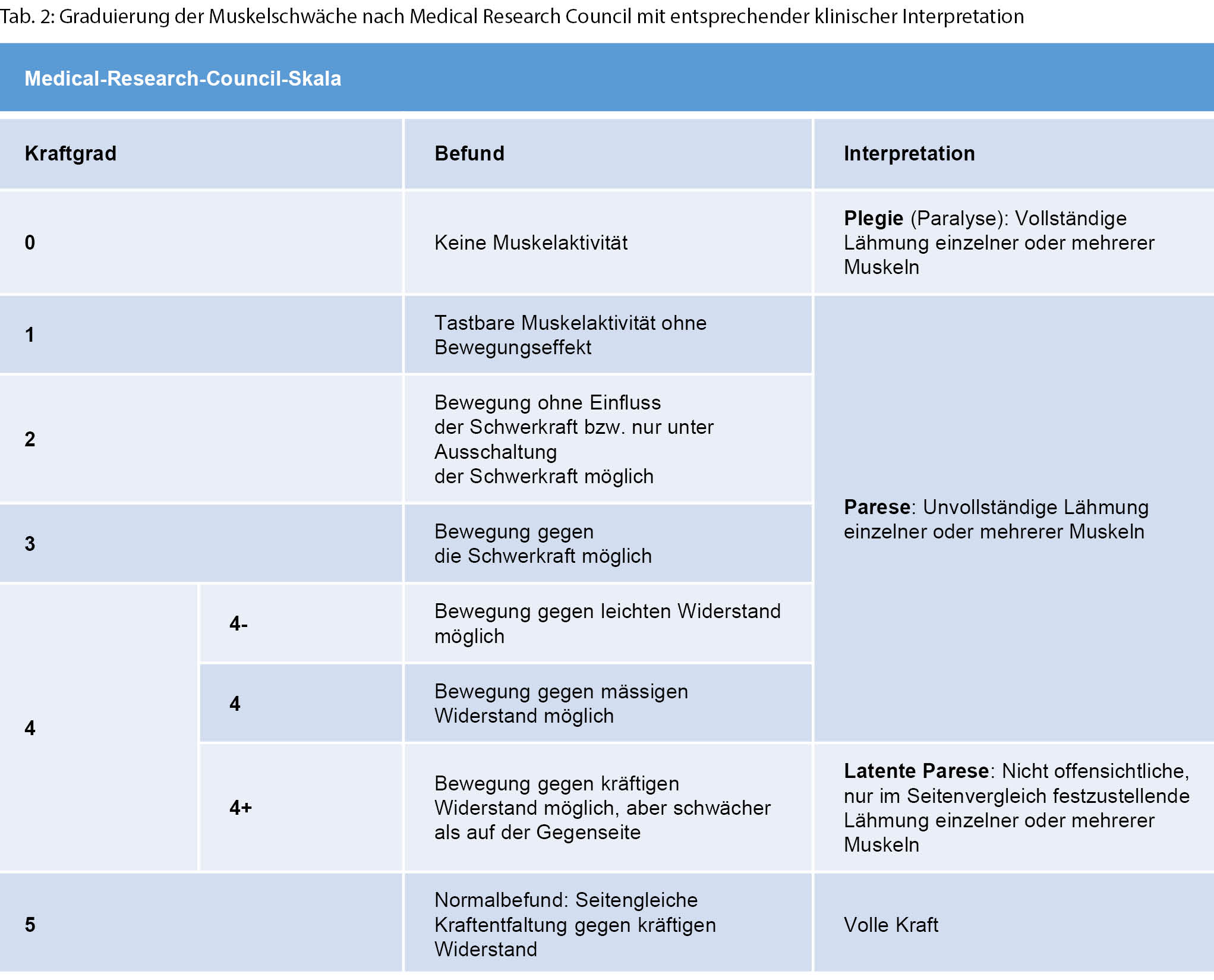
Eine Muskelschwäche wird durch eine formale Krafttestung objektiviert. Hierbei wird die Kraft ermittelt, die notwendig ist, um eine maximale Kraftentwicklung in einem Muskel zu überwinden. Etabliert hat sich hierbei eine Graduierung nach dem Medical Research Council von 0 bis 5 (Tab. 2) [3].
Lokalisation der Muskelschwäche
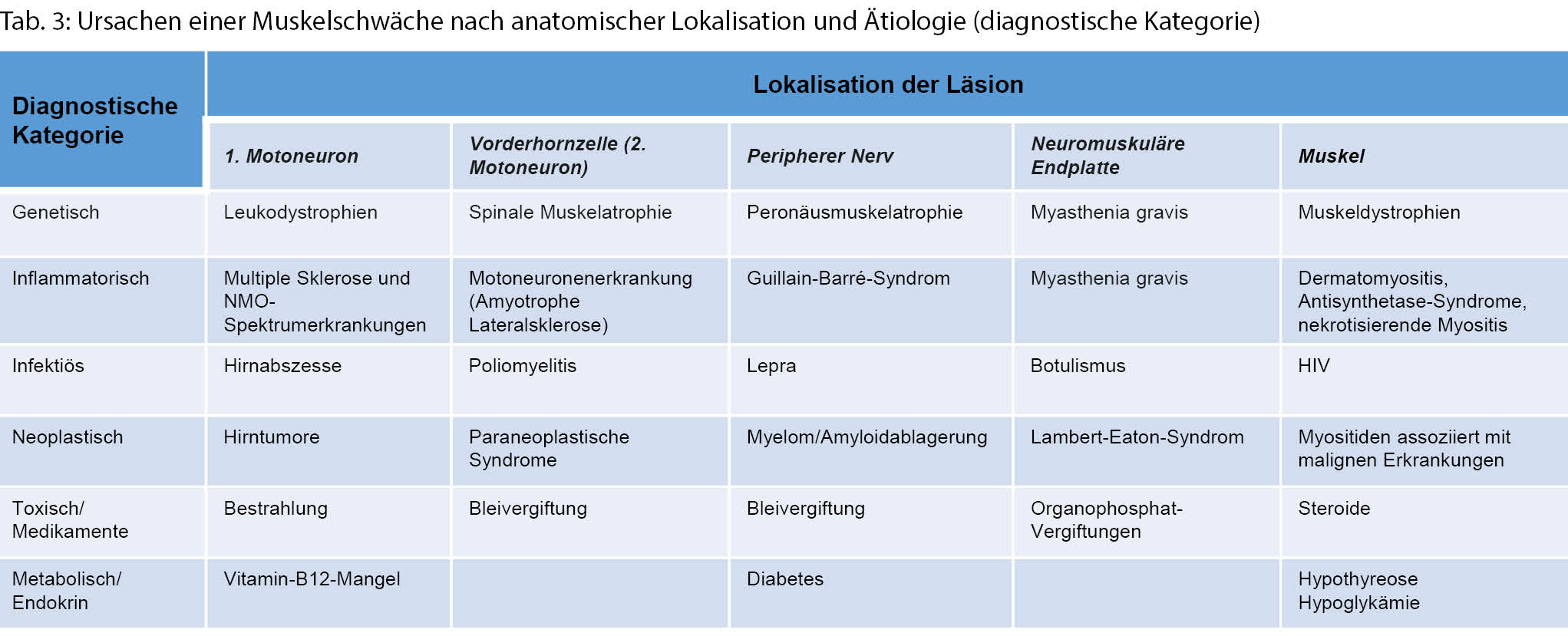
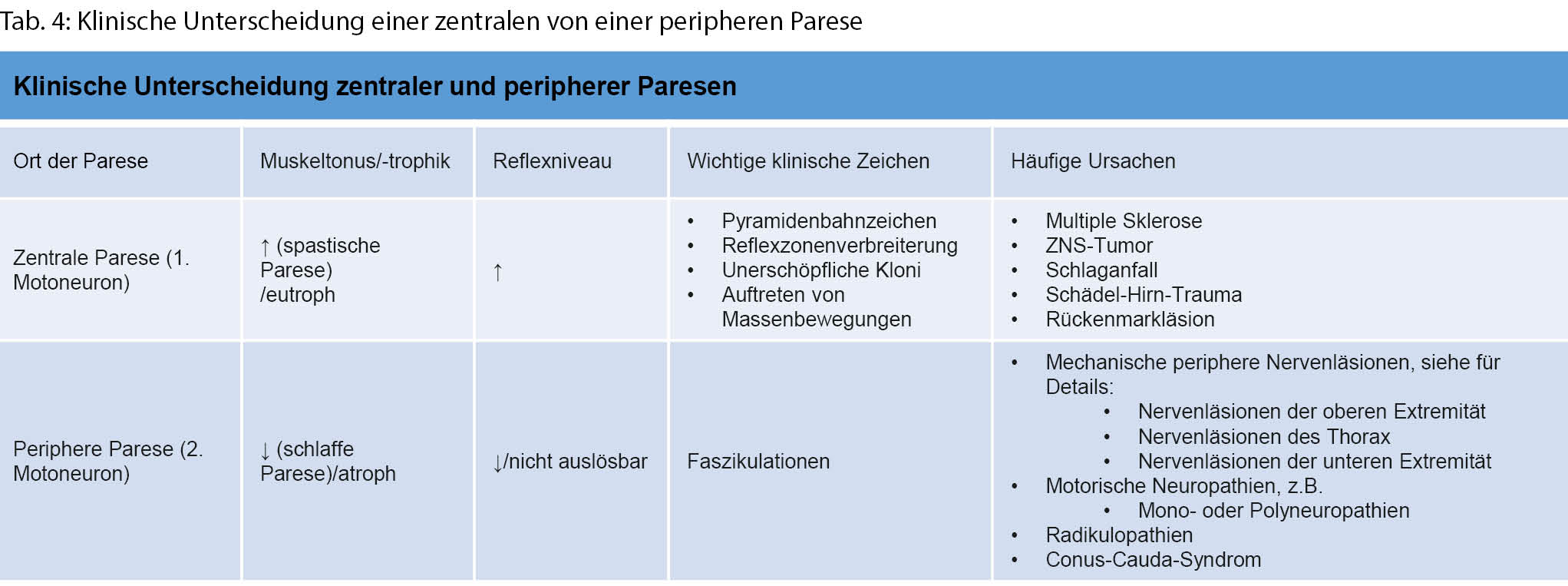
Um die mannigfaltigen Ursachen einer Muskelschwäche genauer eingrenzen zu können, bedarf es einer strukturierten Vorgehensweise und Einordnung gemäss der anatomischen Organisation des neuromuskulären Systems. Von zentral nach peripher beginnt diese mit dem motorischen Kortex und verläuft über den kortikospinalen Trakt, die Motoneurone im Vorderhorn und spinalen Nervenwurzeln zu den peripheren Nerven, der neuromuskulären Endplatte und schliesslich dem Muskel selbst (Tab. 3). Läsionen des zentralen vom peripheren Nervensystems können meistens gut durch eine neurologische Basisuntersuchung abgegrenzt werden (Tab. 4). Eine Untersuchung mittels Elektroneuromyografie (ENMG) kann helfen, unklare Untersuchungsbefunde zu objektivieren.
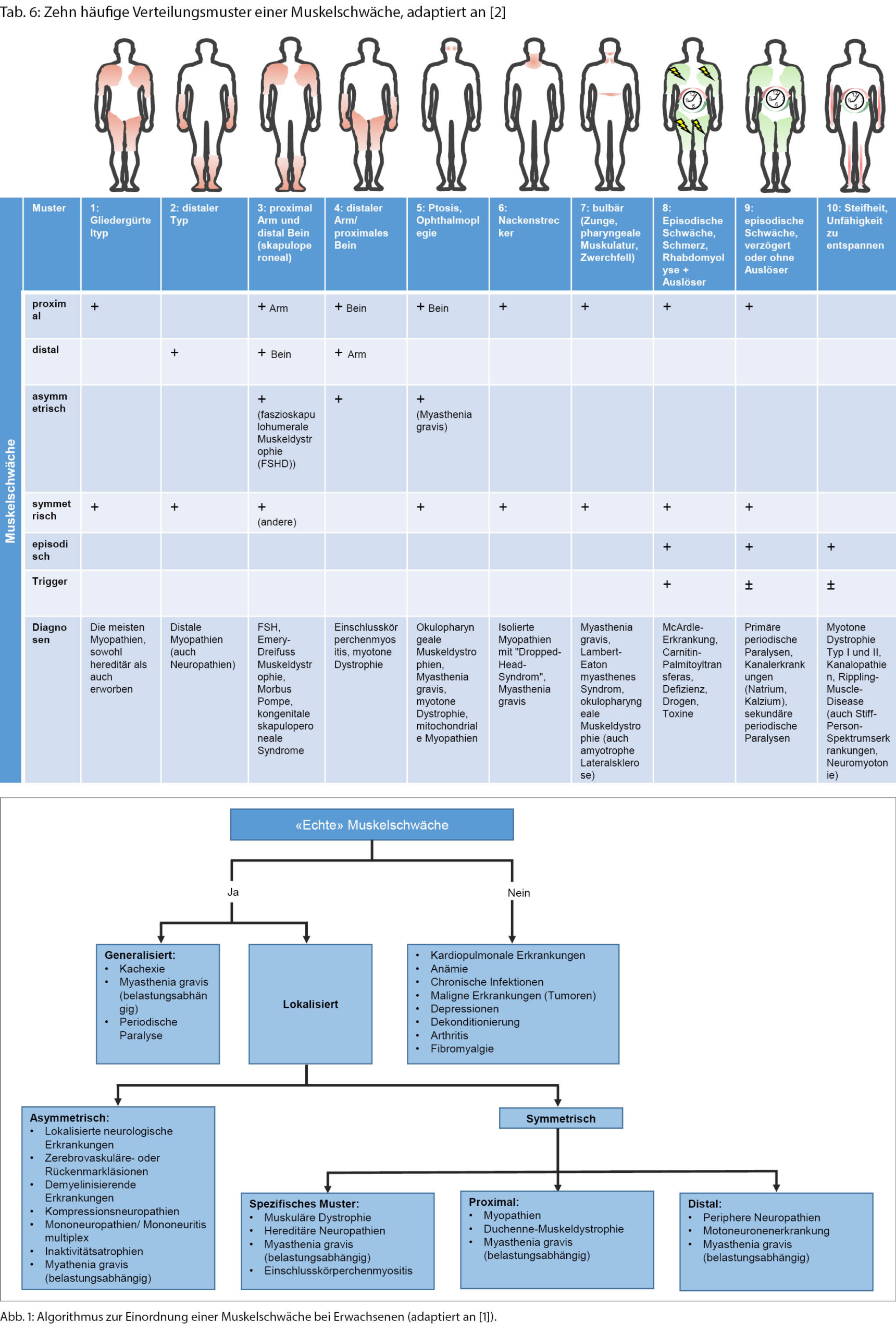
Verteilungsmuster einer muskulären Schwäche als Hilfestellung zur Eingrenzung der Lokalisation und Ursache der Läsion
Wurde eine Muskelschwäche festgestellt, so sollte in einem weiteren Schritt das Verteilungsmuster beschrieben werden, um einer ätiologischen Diagnosestellung näher zu kommen (Abb. 1) [6].
◆ Eine generalisierte muskuläre Schwäche besteht bei den generalisierten Formen der Myasthenia gravis, länger bestehenden periodischen Paralysen (seltenen genetischen Ionenkanalerkrankungen), nach längerer Bettlägerigkeit, Muskelatrophie bei maligner Grunderkrankung oder fortgeschrittener Motoneuronenerkankung.
Wenn die Schwäche nicht generalisiert ist, sollte als nächster Schritt beschrieben werden, ob die Schwäche symmetrisch oder asymmetrisch ausgeprägt ist. Eine asymmetrische Muskelschwäche ist in der Regel durch eine Erkrankung des zentralen oder peripheren Nervensystems bedingt. Zudem haben Läsionen des motorischen Kortex, des Rückenmarks und der spinalen Nervenwurzeln und peripheren Nerven allesamt charakteristische Verteilungsmuster. Eine symmetrische Schwäche kann weiter in ein distales, proximales oder spezifisches Verteilungsmuster aufgeteilt werden [1]:
◆ Eine distale Schwäche ist durch einen reduzierten Faustschluss, eine Schwäche der Handflexion und/oder -extension, reduzierte Plantarflexion und/oder Fussheberschwäche gekennzeichnet. Diese Patientinnen und Patienten haben Probleme auf den Fersen (Steppergang) oder Zehen zu laufen. Eine distal-symmetrische Schwäche ist viel öfter [7] durch eine periphere Neuropathie (v.a. Polyneuropathie) oder beginnende Motoneuronenerkrankung als durch eine Myopathie bedingt (wobei es seltener auch distale Myopathieformen gibt, wie zum Beispiel eine Einschlusskörperchenmyositis mit reduzierter Fingerflexion).
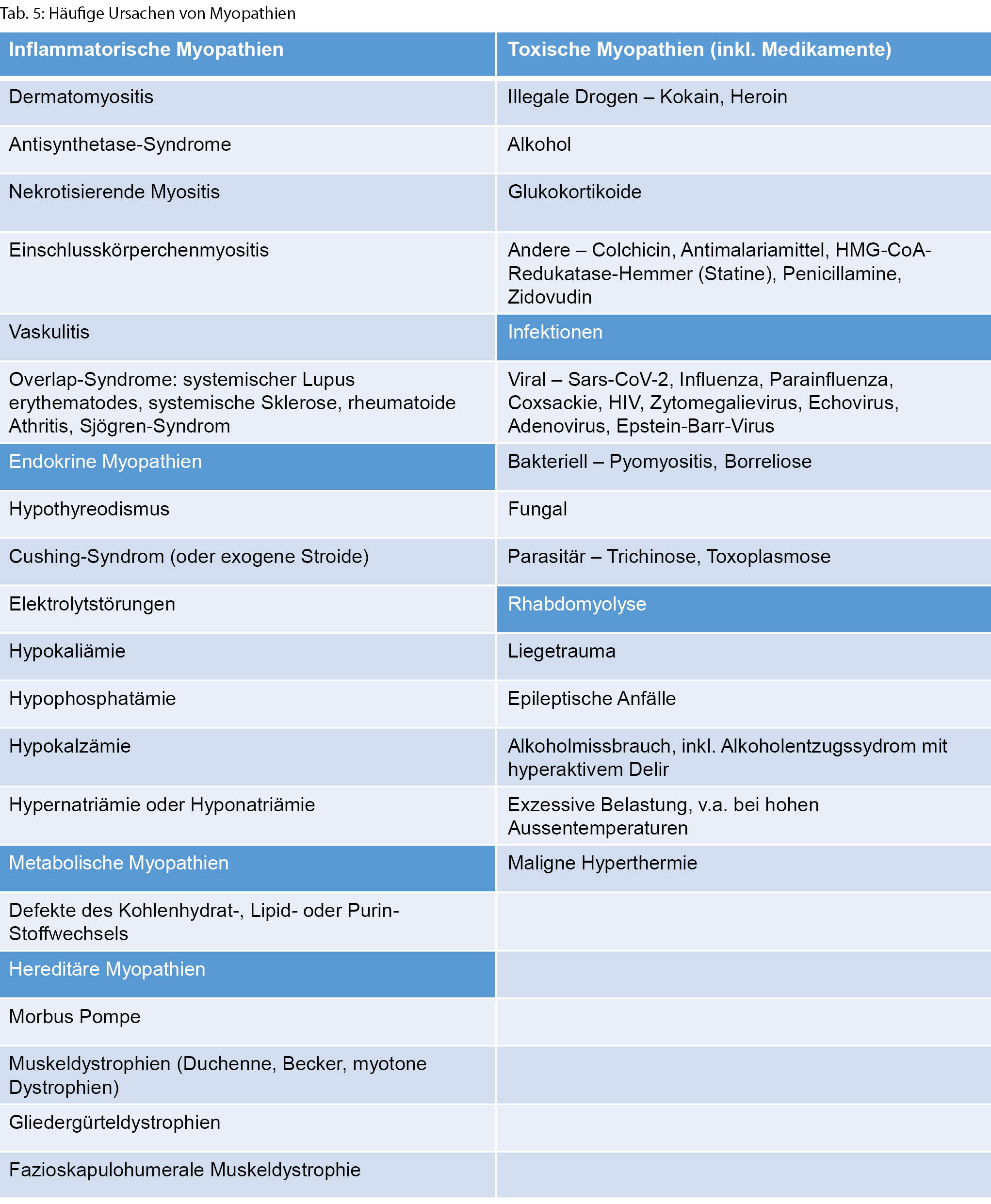
◆ Eine proximale Muskelschwäche umfasst dagegen die axiale Muskulatur, die Schultergürtelmuskulatur und die Hüftflexoren. Betroffene können auch eine Schwäche der Kopfflexion und/oder -extension aufweisen. Eine einfache Möglichkeit, die Funktion der axialen Muskulatur zu überprüfen, ist es, die Betroffenen aus der Rückenlage aufstehen zu lassen und zu beobachten. Bei einer Nackenbeugeschwäche bleibt der Kopf beim Aufsitzen zurück, bei einer Schwäche der Schultergürtelmuskulatur werden die Arme vermindert eingesetzt, und bei einer Schwäche der Bauchmuskulatur wird über eine Seite abgerollt. Die Schwäche der Schultergürtelmuskulatur, insbesondere des M. deltoideus, kann durch Armabduktion mit flektiertem Ellenbogen getestet werden. Es sollte für den Untersucher nicht möglich sein, die Arme herunterzudrücken, wenn die Kraft normal ist. Patientinnen und Patienten mit relevanter Schwäche der vorderen Oberschenkelmuskulatur (M. quadriceps) schaffen es nicht, ohne Hilfe der Arme (verschränkt vor der Brust) aus dem Sitzen aufzustehen oder eine Kniebeuge zu machen. Kompensatorisch wird mit breiter Standachse an «den Beinen entlangkletternd» aufgestanden (Gowers-Zeichen). Eine proximale Muskelschwäche kann bei vielen Myopathien, Muskeldystrophien und der Myasthenia gravis gesehen werden (Tab. 5).
◆ Einige Neuropathien und Myopathien bzw. Muskeldystrophien weisen charakteristische Verteilungsmuster auf. Einige der häufigeren Ursachen bei Erwachsenen sind hier beispielsweise die Einschlusskörperchenmyositis, die Fazioskapulohumerale Muskeldystrophie (FSHD), der Morbus Pompe und die Charcot-Marie- Tooth(CMT)-Neuropathie (Tab. 6).
Ätiologische Einordnung
Ist die Muskelschwäche nun lokalisert und das Verteilungsmuster beschrieben, kann anhand weiterer Untersuchungsbefunde eine Kategorisierung zu einer genetischen, inflammatorischen/immunologischen, infektiösen, neoplastischen, toxischen oder metabolischen Ursache geschehen (Tabelle 3).
Läsionsort
Erkrankungen des 1. Motoneurons
Läsionen des 1. Motoneurons treten nach Schlaganfällen oder bei raumfordernden Läsionen des ZNS oder Rückenmarks auf. Weitere Ursachen vor allem auch des Rückenmarks sind Traumata, Infektionen, Tumore, vaskuläre Anomalien, hypertrophe degenerative skelettale Veränderungen, demyelinisierende Läsionen und angeborene Leukodystrophien. Entsprechende bildmorphologische Untersuchungen und ggf. Liquoranalysen können hier helfen, differenzialdiagnostisch die zugrundeliegende Ätiologie besser abzugrenzen.
Erkrankungen der Vorderhornzellen
Diese treten bei Motoneuronenerkrankungen, der spinalen Muskelatrophie, Bleivergiftungen oder Poliomyelitis auf. Seltene infektiologische Ursachen können das West- Nil- oder Echovirus sein. Insbesondere hier können der zeitliche Verlauf, eine Exposition und das Alter der Erstmanifestation der Symptome sowie ggf. auch eine Liquoranalyse in der Differenzialdiagnose weiterhelfen.
Erkrankungen des peripheren Nervensystems
Krankheitsmanifestationen der peripheren Nerven können typischerweise in zwei Formen auftreten:
◆ als symmetrische Manifestation einer Polyneuropathie, vor allem als Folge eines Diabetes. Andere Ätiologien sind verschiedene toxische oder metabolisch bedingte Störungen. Bei nicht-konklusiver Abklärungsollte je nach Befall und Verlauf auch an eine hereditäre Genese gedacht werden;
◆ als Mononeuropathie, am häufigsten ausgelöst durch eine Kompression (Karpaltunnel- oder Sulcus-Ulnaris- Syndrom). Eine Mononeuritis multiplex (asymmetrische, initial stark schmerzhafte Polyneuropathie) kann durch einen Diabetes oder eine Vaskulitis, wie z.B. eine Polyarteriitis nodosa, bedingt sein.
Eine Auswahl von speziellen behandelbaren Erkrankungen des peripheren Nervensystems, die mit einer Muskelschwäche einhergehen kann, findet sich in Tabelle 7.
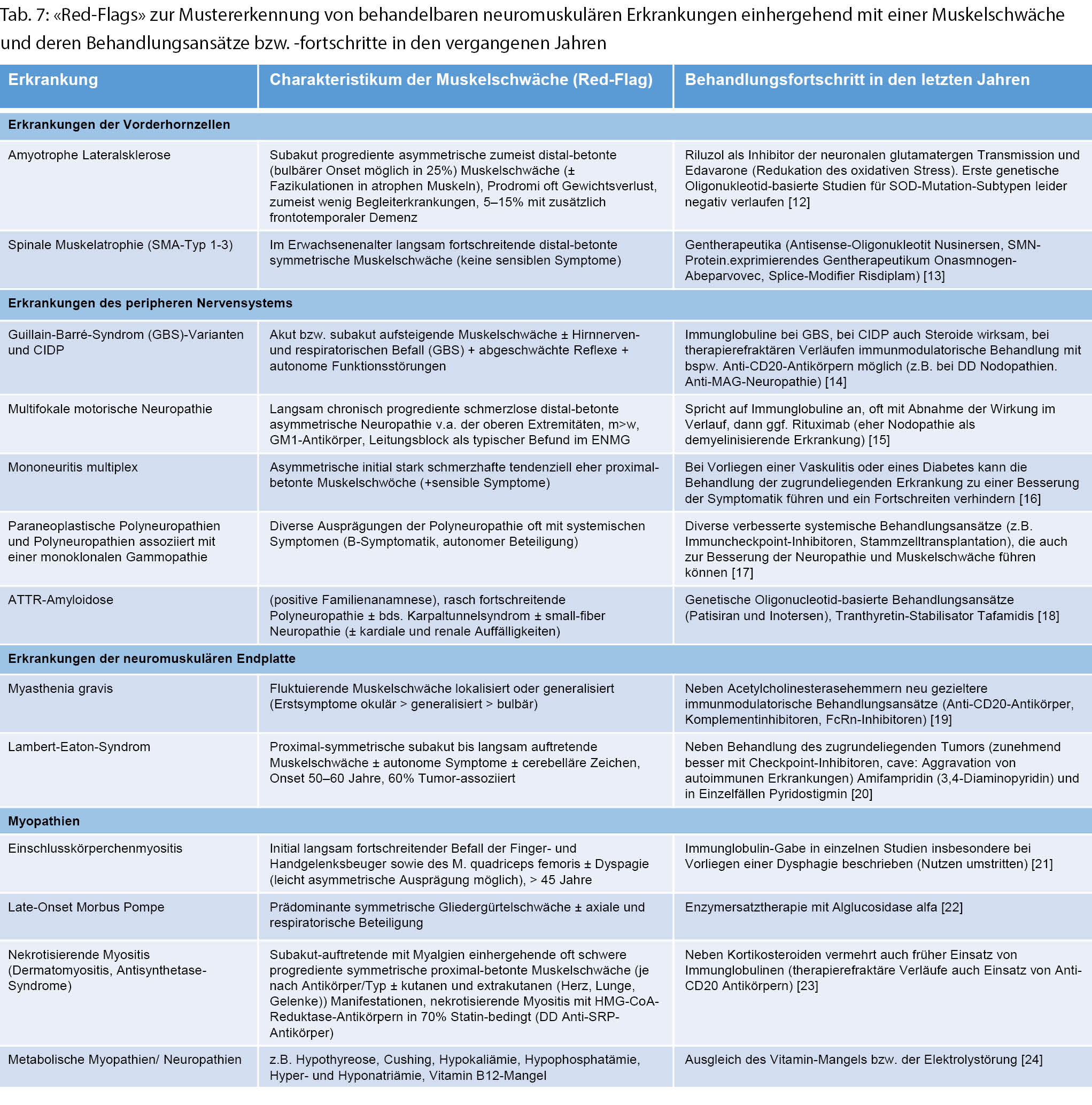
Erkrankungen der neuromuskulären Endplatte
Neben der klassischen autoimmun bedingten Myasthenia gravis können auch eine Inhibition der Acetylcholinesterase durch Organophosphate (Intoxikation) und eine oft paraneoplastisch bedingte Störung der präsynaptischen Kalziumkanäle im Rahmen eines Lambert-Eaton-Syndroms bei einem kleinzelligen Lungenkarzinom eine fluktuierende belastungsabhängige Schwäche bzw. erhöhte muskuläre Ermüdbarkeit/Fatigue bedingen, die für Störungen im Bereich der neuromuskulären Endplatte charakteristisch ist. Hierbei sollte man eine Myasthenie aufgrund ihrer potenziell bedrohlichen Beteiligung der Atem- und Schluckmuskeln (Krise) und Behandelbarkeit nicht verpassen (siehe Abb. 1, Merksatz: «Sag niemals nie zur Myasthenie»). Die Krankheitsausprägung reicht hier von mild und fokal bis zur Generalisierung mit Tetraparese. Neben der klassischen okulären Erstmanifestation (> 50 % der Fälle mit fluktuierender, oft asymmetrischer Ptosis und Doppelbildern) [10], sollte vor allem auch bei fluktuierenden bulbären Symptomen und bei älteren Menschen an eine Myasthenie gedacht werden. Beispiele hierfür sind ein hypophones nasales Sprechen oder Lispeln, das sich mit zunehmender Sprechdauer verschlechtert, oder eine Kauschwäche und -ermüdung, insbesondere für harte Konsistenzen. Oft gehen diese Symptome auch mit einer Dysphagie einher. Eine einfache klinische Testung besteht in der Detektion einer Hypophonie beim laut zählen nach tiefer Inspiration bis bspw. 20 (zwei Zahlen je Sekunde) oder einem Räuspern und Husten nach Trinken eines Glases Wasser als Zeichen für eine Dysphagie. Da die bulbäre Muskulatur sich eine segmentale Innervation mit der respiratorischen Muskulatur teilt, ist die Kenntnis dieser heutzutage sehr gut behandelbaren Krankheitsmanifestationen der Myasthenie wichtig, um Folgebeschwerden wie z.B. einer Mangelernährung und respiratorischen Schwäche mit Gefahr von Aspirationen und Pneumonie früh entgegenwirken zu können.
Myopathien
Die Hauptkategorien von Myopathien, die eine inflammatorische, endokrine, metabolische, toxische oder hereditäre Genese umfassen, sind, wie auch häufige Ursachen einer Rhabdomyolyse, in Tabelle 5 dargestellt.
◆ Alter und Geschlecht können differenzialdiagnostisch helfen, wenn eine Muskeldystrophie vermutet wird.
◆ Eine Anamnese mit wiederholter belastungsabhängiger Pigmenturie (colafarbener Urin) und begleitender Schwäche kann den Verdacht auf eine metabolische Myopathie lenken. Nichtsdestotrotz haben viele metabolische Myopathien aber einen chronisch-progredienten anstatt eines episodischen Verlaufs.
◆ Eine Anamnese der Medikation (z.B. Neuaufnahme Statine, Immuncheckpoint-Inhibitoren), des Alkohol-und Drogenkonsums ist differenzialdiagnostisch unerlässlich.
◆ Einige Endokrinopathien (Hypo- oder Hyperthyreose, Elektrolytstörungen) können bei oft weiteren systemischen Untersuchungsbefunden durch ein einfaches Basislabor eingeordnet werden. An ein Cushing-Syndrom und Steroid-induzierte Myopathie sollte zudem bei proximal betonter Beinmuskelschwäche gedacht werden.
◆ Eine inflammatorische Myopathie kann bei einer proximal betonten symmetrischen Muskelschwäche vermutet werden, wenn alternative Erklärungen für eine Myopathie nicht konklusiv sind und/oder Zeichen für das Vorliegen einer systemischen rheumatolgischen Affektion mit Ausschlag, interstitieller Lungenerkrankung, Polyathritis oder Raynaud-Syndrom bestehen. Der Onset ist hier zumeist mit subakutem Beginn, oft schwererprogredienter symmetrischer, proximal betonter Muskelschwäche und begleitenden Myalgien.
Klinische Untersuchungen
Laborbefunde
Einige Laboruntersuchungen können bei der Evaluation einer Muskelschwäche sinnvoll sein [4].
Klinische Chemie und Urinanalyse: Erhöhungen der Muskelenzyme im Serum (neben der Creatinkinase (CK) auch Aldolase, Laktatdehydrogenase und Aminotransferase) sind suggestiv für das Vorliegen einer Muskelerkrankung. Bei fehlenden klinischen muskulären Symptomen und Untersuchungsbefunden ist aber insbesondere eine Hyper-CK-ämie bis zum 3–5 Fachen der Norm ein weitgehend unspezifischer Laborparameter, der zudem durch vorherige Muskelbelastung, falsch-hohe Werte im Rahmen einer Makro-CK-Ämie, Injektionen in die Muskulatur und Muskeltraumata verzerrt sein kann. Diese Fehlerquellen sollten daher vor der Abnahme einer CK ausgeschlossen werden (drei Tage zuvor keine schwere Muskelbelastung) und teils deutlich voneinander abweichende Normwerte müssen je nach Geschlecht, Ethnie und Alter beachtet werden [11]. Zudem sollte ein Basislabor mit Abnahme u.a. von TSH, Nierenparametern inkl. der Elektrolyte und wichtigsten Vitamine (Folsäure, Vitamin B12 inkl. Methylmalonsäure) erfolgen. Ein blutiger bzw. colafarbener Urin ohne Nachweis von erhöhten Erythrozyten im Urin-Stix ist suggestiv für das Vorliegen einer Myoglobinurie und damit einer Rhabdomyolyse.
Immunologische Parameter sind erforderlich, wenn eine Myasthenie (Anti-AChR, Anti-MuSK, selten Anti- LRP4), inflammatorische Ursache (z.B. Anti-histidyl-t- RNA Synthase [anti-Jo-1], Anti-HMGCR, Anti-SRP) oder Bindegewebserkrankung (Anti-Ro/SSA, Anti-La/SSB, Anti-Sm, und Anti-RNP) vermutet wird.
Anti-neutrophil zytoplasmatische Antikörper(ANCA)- Titer, Hepatitis-B- und C-Serologien und Kryoglobuline können bei Vd.a. das Vorliegen einer systemischen Vaskulitis mit Muskelbeteiligung bestimmt werden. Diese dann zu den inflammatorischen Myopathien gezählte Manifestation ist insgesamt selten und kommt nach einem kürzlich erschienenen Review v.a. im Rahmen einer Polyarteriitis nodosa oder ANCA-assoziierten Vaskulitis vor [9].
Elektrophysiologische Untersuchung
Ein ENMG kann zusammen mit den laborchemischen Resultaten helfen, klinische Befunde weiter differenzialdiagnostisch einzuordnen, insbesondere, wenn klinische Hinweise für den Befall des peripheren Nervensystems, einer neuromuskulären Übertragungsstörung oder eine Myopathie bestehen. Zunehmenden Wert gewinnt hier auch die gezielte Ergänzung mit einem Nerven- bzw. Muskelultraschall (beispielsweise für die Lokalisation
einer Muskelbiopsie oder zugrundeliegender struktureller Ursachen) [8].
Magnetresonanztomografie (MRI)
Die Magnetresonanztomografie gewinnt aufgrund der immer besseren Auflösungsmöglichkeiten und spezifischen Sequenzen als Ganzkörper-MRI bei Muskelerkrankungen und als MRI-Neurografie bei Plexo- oder Neuropathien (insbesondere für im ENMG schwer zu messendem oft proximale Nervenabschnitte (z.B. Plexus-MRI)) einen zunehmenden Stellenwert. Aus den ersichtlichen Veränderungen (z.B. Ödembildung, fibrotischer Umbau) und den Befallsmustern der Muskeln kann zum Beispiel eine gezielte Muskelbiopsie besser geplant und bei Verlaufsuntersuchungen ein Krankheitsprogress bzw.- ansprechen besser abgeschätzt werden. Eine MRI-Untersuchung ohne klinische Befunde ist dagegen bei unspezifischer Muskelschwäche in aller Regel nicht wegweisend [7].
Muskelbiopsie
Die Muskelbiopsie hat in den vergangenen Jahren durch die zunehmenden Möglichkeiten einer genetischen Untersuchung aufgrund der Invasivität der Untersuchungstechnik und Problematik «unspezifischer» Befunde (verschiedene hereditäre oder erworbene Ätiologien können ein ähnliches Muster haben) und «Sampling Errors» an Wertigkeit verloren. Dennoch gibt es vor allem bei den inflammatorischen Nerven- und Muskelerkrankungen (vaskulitische Neuropathien, auch Lymphomatose; Dermatomyositis, Antisynthetase-Syndrome, nekrotisierende immunvermittelte Myositis) sowie bei bestimmten Muskeldystrophien, der Einschlusskörperchenmyositis und Vaskulitiden charakteristische Befunde, die bei der Differenzialdiagnose helfen können. Spezielle Färbungen und enzymatische Testungen können zudem bei der ätiologischen Aufarbeitung bei Vd.a. das Vorliegen von Mitochondriopathien oder weiteren metabolischen Myopathien helfen.
Genetische Testung
Nicht zuletzt aufgrund der erfreulicherweise zunehmenden Behandlungsmöglichkeiten – vor allem auch der genetischen – von Erkrankungen, die mit einer Muskelschwäche einhergehen (Morbus Pompe, SMA, ATTR-Amyloidose) wird die genetische Testung zunehmend früher und parallel in der diagnostischen Aufarbeitung eingesetzt. Neben den therapeutischen Möglichkeiten (Tabelle 6) hat sie ihren Wert auch in der genetischen Beratung, der Prognoseabschätzung sowie der Möglichkeit zur Teilnahme an Studien, die in den vergangenen Jahren exponentiell zugenommen haben.
Respiratorische Muskelschwäche
Wie bereits im Abschnitt der Erkrankungen an der neuromuskulären Endplatte beschrieben, sollte bei Patientinnen und Patienten mit Leitsymptom einer Muskelschwäche die Untersuchung der axialen und respiratorischen Muskulatur nicht vergessen werden, dies insbesondere, wenn zudem eine Dysphagie, Dysarthrie und/oder Dyspnoe besteht.


Universitätsspital Zürich
Frauenklinkstrasse 26, 8048 Zürich
kaimichael.schubert@usz.ch
Es bestehen keine Interessenskonflikte.
Historie
Manuskript akzeptiert: 18.01.2023
ORCID
Kai Michael Schubert
https://orcid.org/0000-0003-1438-7544
Bettina Schreiner
https://orcid.org/0000-0001-6983-4124
1. Shefner JM. Approach to the patient with muscle weakness. Post TW, ed. UpToDate. Waltham, MA: UpToDate Inc. http:// www.uptodate.com; letzter Zugriff: 11.11.2022.
2. Pasnoor M, Dimachkie MM. Approach to Muscle and Neuromuscular Junction Disorders. Continuum (Minneap Minn). 2019; 25(6):1536–1563. DOI: 10.1212/CON.0000000000000799.
3. Medical Research Council. Aids to the Examination of the Peripheral Nervous System. Memorandum no. 45. London; Her Majesty’s Stationery Office: 1981.
4. Rosow LK, Amato AA. The Role of Electrodiagnostic Testing, Imaging, and Muscle Biopsy in the Investigation of Muscle Disease. Continuum (Minneap Minn) 2016;22:1787.
5. Glaubitz S, Schmidt K, Zschüntzsch J, Schmidt J. Myalgia in myositis and myopathies. Best Pract Res Clin Rheumatol. 2019;33(3):101433. DOI: 10.1016/j.berh.2019.101433.
6. Barohn RJ, Dimachkie MM, Jackson CE. A pattern recognition approach to patients with a suspected myopathy. Neurol Clin. 2014;32(3):569–93, vii. DOI: 10.1016/j.ncl.2014.04.008.
7. Nicolau S, Naddaf E. Muscle MRI for Neuromuscular Disorders. Pract. Neurol. 2020;27–32.
8. Wijntjes J, van Alfen N. Muscle ultrasound: Present state and future opportunities. Muscle Nerve. 2021;63(4):455–466. DOI: 10.1002/mus.27081.
9. Conticini E, d’Alessandro M, Al Khayyat SG, et al. Inflammatory muscle involvement in systemic vasculitis: A systematic review. Autoimmun Rev. 2022;21(3):103029. DOI: 10.1016/j.aut rev.2021.103029.
10. Beloor Suresh A, Asuncion RMD. Myasthenia Gravis. In: StatPearls [Internet]. Treasure Island (FL); StatPearls: 2022.
11. Kley RA, Schmidt-Wilcke T, Vorgerd M. Differential Diagnosis of HyperCKemia. Neurology Internat Open. 2018; 02(01): E72– E83, DOI: 10.1055/s-0043-124361.
12. Quinn C, Elman L. Amyotrophic Lateral Sclerosis and Other Motor Neuron Diseases. Continuum (Minneap Minn). 2020; 26(5):1323–1347. DOI:10.1212/CON.0000000000000911
13. Nance JR. Spinal Muscular Atrophy. Continuum (Minneap Minn). 2020;26(5):1348–1368. DOI: 10.1212/CON.0000000000 000918.
14. Van den Bergh PYK, van Doorn PA, Hadden RDM, et al. European Academy of Neurology/Peripheral Nerve Society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint Task Force-Second revision [published correction appears in J Peripher Nerv Syst. 2022;27(1):94] [published correction appears in Eur J Neurol. 2022;29(4):1288]. J Peripher Nerv Syst. 2021;26(3):242–268. DOI: 10.1111/jns.12455.
15. Beadon K, Guimarães-Costa R, Léger JM. Multifocal motor neuropathy. Curr Opin Neurol. 2018;31(5):559–564. DOI: 10. 1097/WCO.0000000000000605.
16. Karam C. Peripheral Neuropathies Associated With Vasculitis and Autoimmune Connective Tissue Disease. Continuum (Minneap Minn). 2020;26(5):1257–1279. DOI: 10.1212/CON.00000 00000000917.
17. Antoine JC, Camdessanché JP. Paraneoplastic neuropathies. Curr Opin Neurol. 2017;30(5):513–520. DOI: 10.1097/WCO.000 0000000000475.
18. Adams D, Ando Y, Beirão JM, et al. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol. 2021;268(6):2109–2122. DOI: 10.1007/s00415–019–09688–0.
19. Narayanaswami P, Sanders DB, Wolfe G, et al. International Consensus Guidance for Management of Myasthenia Gravis: 2020 Update. Neurology. 2021;96(3):114–122. DOI: 10.1212/ WNL.0000000000011124.
20. Jayarangaiah A, Theetha Kariyanna P. Lambert Eaton Myasthenic Syndrome. In: StatPearls. Treasure Island; StatPearls: 2022.
21. Goyal NA. Inclusion Body Myositis. Continuum (Minneap Minn). 2022;28(6):1663–1677. DOI: 10.1212/CON.0000000000001 204.
22. Morales A, Anilkumar AC. Glycogen Storage Disease Type II. In: StatPearls. Treasure Island (FL;: StatPearls: 2022.
23. Manousakis G. Inflammatory Myopathies. Continuum (Minneap Minn). 2022;28(6):1643–1662. DOI: 10.1212/CON.000000 0000001179.
24. Tarnopolsky MA. Metabolic Myopathies. Continuum (Minneap Minn). 2022;28(
PRAXIS
- Vol. 112
- Ausgabe 9
- Juli 2023