Der 27-jährige Patient stellte sich aufgrund von einseitigen Beinschmerzen vor. In der Vorgeschichte war ein Diabetes insipidus respektive Panhypopituitarismus bekannt. Laboranalytisch bestand eine unzureichende Hormonsubstitution. Im MRI fiel eine grosse Kontrastmittel-aufnehmende Raumforderung in der Hypophyse mit Ausdehnung bis in den Hypothalamus auf. Mittels FDG-PET/CT konnte eine hypermetabole Läsion im Bereich des Femurschaftes links dargestellt werden. Nach Biopsie der Läsion konnte die Diagnose einer multisystemischen Langerhans-Zell-Histiozytose gestellt werden.
Anamnese und Befunde
Der 27-jährige Patient stellte sich aufgrund von Oberschenkelschmerzen linksseitig vor. Die Beschwerden hätten seit einigen Wochen bestanden und an Intensität zugenommen. Die Schmerzen seien in Ruhe von dumpfer Qualität, bei Belastung stechend mit Ausstrahlung in das Knie. Der Patient betreibt regelmässig Kampfsport und fühlte sich durch die Schmerzen eingeschränkt. Die eingenommenen Schmerzmittel (Paracetamol, Ibuprofen) hätten nicht geholfen.
Als Vorerkrankung wurde beim Patienten im Alter von 15 Jahren ein Diabetes insipidus diagnostiziert. Die Abklärungen wurden aufgrund einer zunehmenden Schwäche mit begleitendender Polyurie und Polydipsie veranlasst. Im damals durchgeführten MRI des Neurocraniums wurde ein verdickter Hypophysenstiel mit diffuser Kontrastmittelanreicherung der Hypophyse festgestellt. Das Ganzkörper MRI war unauffällig. Es wurde ein Diabetes insipidus am ehesten im Rahmen einer lymphozytären Hypophysitis festgehalten. Einige Jahre nach Diagnosestellung entwickelte der Patient weitere Hormonausfälle (Hypocortisolismus, Hypothyreose, Hypogonadismus, Wachstumshormonmangel), so dass retrospektiv ein Panhypopituitarismus diagnostiziert wurde. Der Patient hatte allerdings keine regelmässigen endokrinologischen Kontrollen, sodass bislang nur eine Behandlung des Diabetes insipidus mit Desmopressin erfolgte und die übrigen Hormonachsen nicht substituiert wurden.
Systemanamnestisch lagen keine B-Symptome vor. Seit einigen Jahren bestand vermehrte Müdigkeit, keine Visusstörungen und kein regelmässiger Alkohol- oder Nikotinkonsum.
Im Status wies der Patient einen adipösen Habitus (BMI 34 kg/m2) mit fahlem Hautkolorit und spärlicher Körperbehaarung auf. Die klinische Untersuchung (inklusive Hirnnervenstatus) war unauffällig. Im Bereich der beklagten Schmerzen am linken Bein war keine Hautrötung, keine Druckdolenz oder Überwärmung festzustellen und die Untersuchungen von Hüfte und Knie waren blande. Laboranalytisch fanden sich bis auf den Panhypopituitarismus mit inadäquater Hormonsubstitution keine Auffälligkeiten, insbesondere keine erhöhten Entzündungswerte und keine Blutbildveränderungen.
Zusammenfassend handelt es sich um einen 27-jährigen Patienten mit einseitigen Beinschmerzen und einem langjährig vorbekannten, nicht adäquat substituierten Panhypopituitarismus mit Diabetes insipidus in der Annahme einer durchgemachten lymphozytären Hypophysitis.
Differenzialdiagnostische Überlegungen
Muskuloskelettale Beschwerden sind ein häufiges Problem im klinischen Alltag. (Post)traumatische Ursachen, meist im Bereich von Gelenken, sind bei jungen, respektive aktiven Patienten die häufigste Ursache. Eine solche Anamnese liegt jedoch bei unserem Patienten nicht vor. Bei atypischer Lokalisation und länger anhaltenden unerklärten Knochenschmerzen, muss auch an eine neoplastische Genese gedacht werden. Bei jungen Patienten sind das in erster Linie primäre Knochentumore, bei älteren Patienten sind Metastasen (Prostata-, Mamma-, Bronchialkarzinom) oder Manifestationen eines Multiplen Myeloms zu erwarten.
Im Gegensatz zu den Knochenschmerzen ist ein zentraler Diabetes insipidus, respektive ein Panhypopituitarismus sowohl bei Kindern als auch bei Erwachsenen ein sehr seltenes Krankheitsbild. Die häufigste Ursache des zentralen Diabetes insipidus ist idiopathisch. Bekannte Auslöser sind primäre Tumore (meist Kraniopharyngeom) oder sekundäre intrakranielle Neoplasien (Metastasen, Lymphome, Langerhans-Zell-Histiozytose (LCH)), infiltrative oder entzündliche Erkrankungen (Sarkoidose, Granulomatose mit Polyangiitis, autoimmune lymphozytäre Hypophysitis) oder traumatische Ursachen (Fraktur, neurochirurgischer Eingriff) [1] (Tabelle1). Bei allen Formen können magnetresonanztomographisch unspezifische Veränderungen (Verdickung des Hypophysenstiels, gesteigerte Kontrastmittel-Anreicherung) auftreten, so dass die bildmorphologischen Veränderungen bezüglich der Diagnosefindung oft nicht weiterhelfen. Aufgrund der Lokalisation ist die Abklärung mittels Gewebeuntersuchung eingeschränkt und oft nicht vertretbar.

Weitere Abklärungsschritte und Verlauf
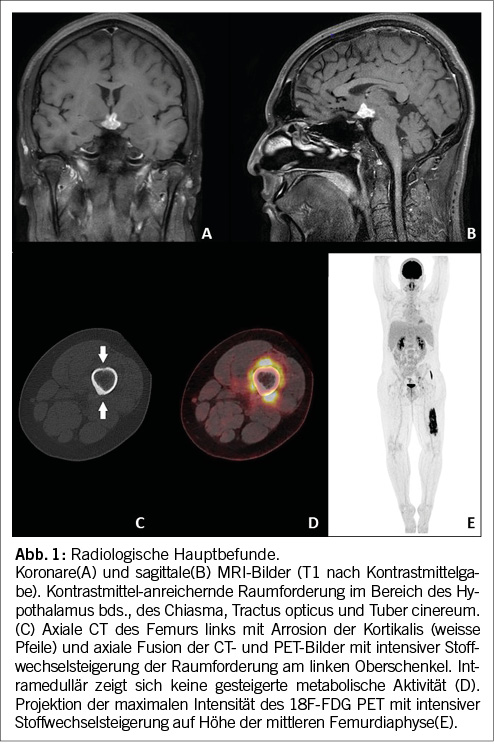
Aufgrund des nicht adäquat substituierten Panhypopituitarismus erfolgte zur Standortbestimmung ein MRI des Neurocraniums. Hier konnte eine atrophierte Hypophyse mit fadendünnem Hypophysenstiel und fehlendem Hypophysenhinterlappen-Signal dargestellt werden. Des Weiteren bestand eine deutlich Kontrastmittel-aufnehmende Raumforderung im Bereich des Hypothalamus, des Chiasmas und Tractus opticus beidseits (Abb.1A und B).
Diese Befunde eines jungen Patienten mit Diabetes insipidus mit progredientem Hormonausfall, atrophierter Hypophyse mit fadendünnem Hypophysenstiel und deutlich Kontrastmittel aufnehmender Raumforderung im Hypothalamus und atraumatischen Knochenschmerzen passen zu einer Manifestation einer multisystemischen LCH, weshalb wir im nächsten Schritt ein Ganzkörper 18F-fluorodeoxyglucose (FDG)PET/CT durchführten. Die Untersuchung zeigte eine intensive FDG-Aufnahme in der hypothalamischen Raumforderung. Im Bereich der beklagten Schmerzen (Femurdiaphyse links) fand sich eine intensiv FDG-avide Raumforderung in der Muskulatur rund um die Femurdiaphyse mit lokalen Arrosionen der angrenzenden Kortikalis, ohne gesteigerte Stoffwechselaktivität im Knochenmark (Abb. 1C und D). Diese Läsion am Femur links war gut zugänglich für eine CT-gesteuerte Biopsie.
Diagnose
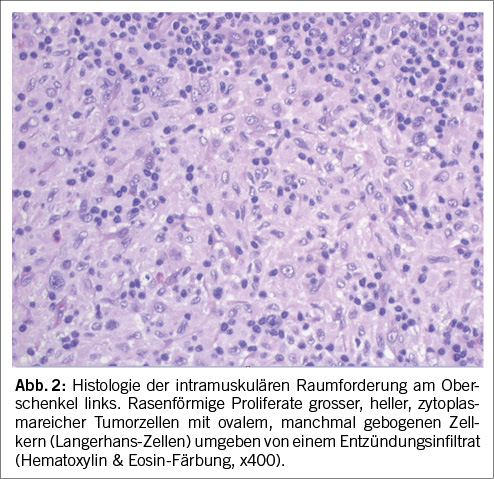
Mittels CT-gesteuerter Biopsie konnte Gewebe perifemoral gewonnen werden. In der histopathologischen Aufarbeitung zeigte sich ein Eosinophilen-reiches Entzündungsinfiltrat mit proliferierten atypischen Zellen mit „Kaffeebohnen-artigen“ Kernen. Die Immunhistochemie mit dem Nachweis der typischen Marker CD1a, Langerin, S100 und CD68 führte zur Diagnose einer LCH (Abb. 2). Somit wurde die Diagnose einer multisystemischen LCH mit Hirn und Weichteilbeteiligung gestellt.
Kommentar
Die LCH gehört zusammen mit der Erdheim-Chester Erkrankung zu den häufigsten histiozytären Erkrankungen, wobei es sich insgesamt um sehr seltene Krankheitsentitäten handelt. Die Inzidenz wird auf 1 Fall/1.5 Millionen Menschen pro Jahr geschätzt [2]. Sowohl Kinder als auch Erwachsene können betroffen sein, mit höherer Inzidenz bei Kindern.
Historisch wurde die LCH als entzündliches Geschehen betrachtet und war auch bekannt unter dem Namen „Histiozytosis X“ oder „Hand-Schüller Christian“ Krankheit. Mittlerweile konnte jedoch gezeigt werden, dass die LCH durch eine unkontrollierte Proliferation von Antigen präsentierenden Zellen, den Langerhans Zellen, entsteht. Heutzutage ist bekannt, dass >50% der LCH Fälle eine BRAF p.V600E Mutation[3] und >90% der LCH/ECD Fälle eine aktivierende Mutation im Mitogen-activated-protein kinase/extracellular-signal-regulated kinase (MAPK/ERK) Signalweg aufweisen[4]. Nach diesen Erkenntnissen wurden die histiozytären Erkrankungen 2017 den hämatopoietischen Neoplasien gemäss WHO zugeordnet [5].
Klinisch handelt es sich um ein sehr heterogenes Krankheitsbild mit unterschiedlichem Verlauf vom radiologischen Zufallsbefund bis zum Multiorganversagen. Grundsätzlich wird zwischen einer unifokalen und multifokalen/multisystemischen Erkrankung mit Mehrorganbeteiligung unterschieden. Am häufigsten manifestiert sich die Krankheit im Knochen, meistens in Form von Osteolysen und in der Hypophyse mit prädominantem Diabetes insipidus, der den weiteren Manifestationen viele Jahre vorausgehen kann. Die LCH der Lunge im frühen Stadium präsentiert sich meist in Form von peribronchialen, pulmonalen Noduli mit Transformation zu Zysten im Verlauf der Erkrankung[6]. Die pulmonale LCH ist meist mit Nikotinkonsum assoziiert und wird als Spezialentität betrachtet [7]. Letztlich können aber alle Organe betroffen sein. Die Beschwerden sind meist unspezifisch, was zu einer Verzögerung der Diagnose über Jahre führen kann.
Bei unserem Patienten wurde im Kindesalter ein Diabetes insipidus festgestellt. Eine Systemerkrankung wurde zum Diagnosezeitpunkt gesucht, jedoch nicht gefunden. Aufgrund der Lokalisation (Hypophyse) wäre eine Biopsie mit nicht vertretbarer Morbidität verbunden gewesen. Wie bei unserem Patienten kommt es bei einem hypophysären Befall der LCH häufig als erstes zu einem Ausfall der Hormone aus dem Hypophysenhinterlappen, im Verlauf zu einem progredienten Hormonausfall aus dem Hypophysenvorderlappen und Jahre später zur Beteiligung weiterer Organe (in unserem Fall Weichteile). Die ossäre Beteiligung der LCH ist relativ häufig, dabei ist ein Weichteilbefall typischerweise die Folge einer Ausbreitung aus dem benachbarten Knochen/Knochenmark. Eine primäre Weichteilbeteiligung mit sekundärer Arrosion der benachbarten Kortikalis, wie bei unserem Patienten, ist in der Literatur selten beschrieben. Die Diagnose einer LCH stützt sich auf den histopathologischen Nachweis der Langerhans-Zell-Infiltrate. Eine Biopsie der Hypophyse wird in der Regel nicht durchgeführt, so dass bei einer Hypophysen Manifestation und Verdacht auf eine LCH ein Ganzkörper FDG-PET/CT die Standarduntersuchung ist[7]. Da LCH Läsionen häufig sehr stark FDG-avide sind, wird versucht die Läsion mit dem stärksten Hypermetabolismus zu biopsieren. Aufgrund der unterschiedlichen Zellularität und Beimischung von Entzündungszellen sind grosszügige Biopsien für eine korrekte Diagnose erforderlich.
Histopathologisch sind LCH Läsionen durch proliferierte Zytoplasma-reiche Zellen mit Kaffeebohnen-artigem Kern mit häufig länglicher Membranfurchung charakterisiert. Je nach Aktivität der Langerhans-Zell-Histiozytose finden sich beigemischte eosinophile Granulozyten unterschiedlicher Dichte sowie Lymphozyten und Plasmazellen. Die diagnostischen immunohistochemischen Marker für Langerhans-Zellen sind CD1a und Langerin (CD207). Da >50% der LCH eine BRAF pV600E Mutation aufweisen [3], ist auch die BRAF-V600E Immunhistochemie diagnostisch hilfreich. Bei fehlender immunhistochemischer Expression von BRAF, respektive negativer Mutationsanalyse wird meist ein NGS für Gene, welche im MAPK-ERK Signalweg involviert sind, durchgeführt [7].
Bei unserem Patienten konnte weder eine BRAF Mutation noch eine Alteration der Gene MAP2K1, KRAS, NRAS oder PIK3CA nachgewiesen werden.
Die therapeutischen Möglichkeiten unterscheiden sich stark. Eine unifokale LCH ist bei Erwachsenen Patienten häufig kurativ behandelbar, wobei es verschiedene lokale Therapien gibt (z.B. Radiotherapie, chirurgische Resektion, Steroidinfiltration). Im Spezialfall der Single-system pulmonalen LCH sollte zwingend ein Rauchstopp empfohlen werden. Dies alleine kann bereits zu einem vollständigen Rückgang der LCH Läsionen führen [7].
Da die Krankheit sehr selten ist und es nur äusserst wenige prospektive Studien gibt, ist der optimale Behandlungsalgorithmus der multisystemischen Krankheit unklar. Bei Patienten mit asymptomatischer Erkrankung und ohne Beteiligung von kritischen Organen (wie Hirn, Leber und Lunge) oder Vorliegen einer Endorgan-Dysfunktion kann vorerst beobachtet werden. Bei Patienten mit symptomatischer Erkrankung oder Beteiligung von Hirn, Leber und Lunge gibt es verschiedene therapeutische Möglichkeiten von konventionellen Chemotherapeutika, Bisphosphonaten (bei Knochen prädominanter Erkrankung), Immunmodulatoren, Hydroxyurea, Methotrexat, Hochdosistherapie mit ASCT und zielgerichteten Therapien wie BRAF-und MEK-Inhibitoren. Aufgrund des schnellen Ansprechens und der hohen Ansprechrate wird bei Befall von kritischen Organen (Hirn, Leber, Milz) eine zielgerichtete Therapie favorisiert [7].
Unser Patient ist sehr jung und hatte einen „kritischen Organbefall“ (Gehirn), sodass wir eine MEK-Inhibitor Therapie mit Cobimetinib empfohlen haben. Nach wenigen Wochen Behandlung war unser Patient schmerzfrei. Im FDG-PET/CT konnte nach drei Monaten eine vollständige metabolische Remission der LCH Manifestation am Femurschaft und eine deutliche Re-gredienz der Läsion im Hypothalamus und Hypophyse festgestellt werden. Nach Einleitung der Substitutionstherapie mit Levothyroxin, Testosteron und Hydrocortison verbesserte sich die Leistungsfähigkeit des Patienten markant. Trotz des guten Therapieansprechens wird der Patient lebenslang auf eine Hormonsubstitutionstherapie angewiesen sein.
Medizinische Onkologie und Hämatologie
Kantonsspital Winterthur
Brauerstrasse
8401 Winterthur
Martina.bertschinger@ksw.ch
Es bestehen keine Interessenskonflikte.
Historie
Manuskript eingereicht: 30.07.2023
Nach Revision angenommen: 18.10.2023
1. Maghnie M, Cosi G, Genovese E, et al. Central diabetes insipidus in children and young adults., N Engl J Med. 2000;343(14):998-1007.
2. Makras P, Stathi D, Yavropoulou M, et al, The annual incidence of Langerhans cell histiocytosis among adults living in Greece. Pediatr Blood Cancer. 2020;67(9):e28422.
3. Badalian-Very G, Vergilio J, Degar B, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116(11):1919-1923.
4. Diamond EL, Durham B, Haroche J, et al. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discov. 2016;6(2):154-165.
5. Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues: International Agency for Research on Cancer, 2017
6. Brauner MW, Grenier P, Tijani K, Battesti JP, Valeyre D. Pulmonary Langerhans cell histiocytosis: evolution of lesions on CT scans. Radiology. 1997;497-502.
7. Goyal G, Tazi A, Go RS, et al., International expert consensus recommendations for the diagnosis and treatment of Langerhans cell histiocytosis in adults, Blood. 2022;139 (17): 2601–2621.
PRAXIS
- Vol. 113
- Ausgabe 1
- Januar 2024