Presentation du cas
Contexte
Il s’ agit d’ un patient de 59 ans, récemment diagnostiqué d’ une maladie de Waldenström. Il est référé dans un centre universitaire pour gestion d’ une diathèse hémorragique et suspicion d’ un syndrome d’ hyperviscosité avec indication à une plasmaphérèse. Les éléments en faveur d’ un syndrome d’ hyperviscosité comprenaient l’ hyperparaprotéinémie, l’ épistaxis et l’ engorgement des veines rétiniennes au fonds d’ œil. Concernant le traitement de la maladie de Waldenström le patient suit un schéma incluant le bortezomib (Velcade®) et dexaméthasone.
Anamnese
A l’ anamnèse, le patient ne reporte pas de symptômes en lien avec un syndrome d’ hyperviscosité, notamment pas de troubles visuels, signes neurologiques ou dyspnée. En revanche, il décrit l’ apparition d’ une épistaxis récidivant depuis environ 2–3 mois ainsi que des ecchymoses d’ apparition spontanée. Il n’ y a pas d’ autres signes d’ une diathèse hémorragique, notamment pas de méléna, hématurie ou saignement prolongé. A noter que le patient a eu une colectomie post-diverticulite en 2019, sans complication hémorragique dans les suites chirurgicales. Il n’ y a pas de pathologie hématologique connue dans la famille.
Status
Au niveau du status, on objective des hématomes aux niveaux des membres, d’ âges différents. Il n’ y a pas de pétéchies ni de purpura. L’ examen ORL ne retrouve pas de saignement. Le status cardiopulmonaire est sans particularité.
Examens Complémentaires
La fonction rénale est conservée, et aucun trouble électrolytique n’ est objectivé. L’ hémogramme retrouve une hémoglobine stable à 74 g/l, avec un hématocrite à 0.23 l/l, des réticulocytes à 17.8 G/l, et des thrombocytes à 126 G/l. Il n’ y a pas d’ éléments en faveur d’ un syndrome de lyse. L’ électrophorèse des protéines met en évidence des protéines totales à 99 g/l avec un pic d’ IgM à 70 g/l. Les tests de la crase démontrent une hémostase altérée avec TP 70 %, INR 1.2, aPTT 51 sec et temps de thrombine 15 sec. Le fibrinogène et les D-dimères sont dans la norme. Le dosage des facteurs de la coagulation retrouve une diminution de l’ activité du facteur VIII (FVIII) coagulant (29 %) et chromogénique, et du facteur von Willebrand (FVW) activé (17 %) et antigénique (20 %).
Diagnostic
Les analyses de coagulation mettent ainsi en évidence un déficit en FVIII et FVW. Le diagnostic différentiel se pose entre une maladie von Willebrand héréditaire et acquise. Au vu de l’ absence d’ antécédent de diathèse hémorragique, et du contexte hémato-oncologique, le diagnostic d’ un déficit acquis secondaire à la paraprotéinémie est suspecté. Le syndrome de l’ hyperviscosité est évoqué au vu d’ une IgM à 70 g/l, d’ une atteinte rétinienne et de l’ épistaxis.
Traitement
L’ indication à la plasmaphérèse, à but d’ épurer la paraprotéine IgM, est double: pour possible syndrome d’ hyperviscosité (niveau d’ évidence 1B) (1) et syndrome de von Willebrand acquise paranéoplasique. La mise en place de l’ accès vasculaire par cathéter de dialyse fémoral est encadrée par un traitement substitutif (Wilate® 3000 UI: FVW et FVIII avec un rapport 1 : 1). Il bénéficie de deux séances de plasmaphérèses avec anticoagulation loco-régionale par citrate et volume plasmatique échangé de 3900 ml par séance composé de 2 culots de plasma frais congelé et 3000 ml d’ albumine 5 % et 500 ml de NaCl 0.9 %.
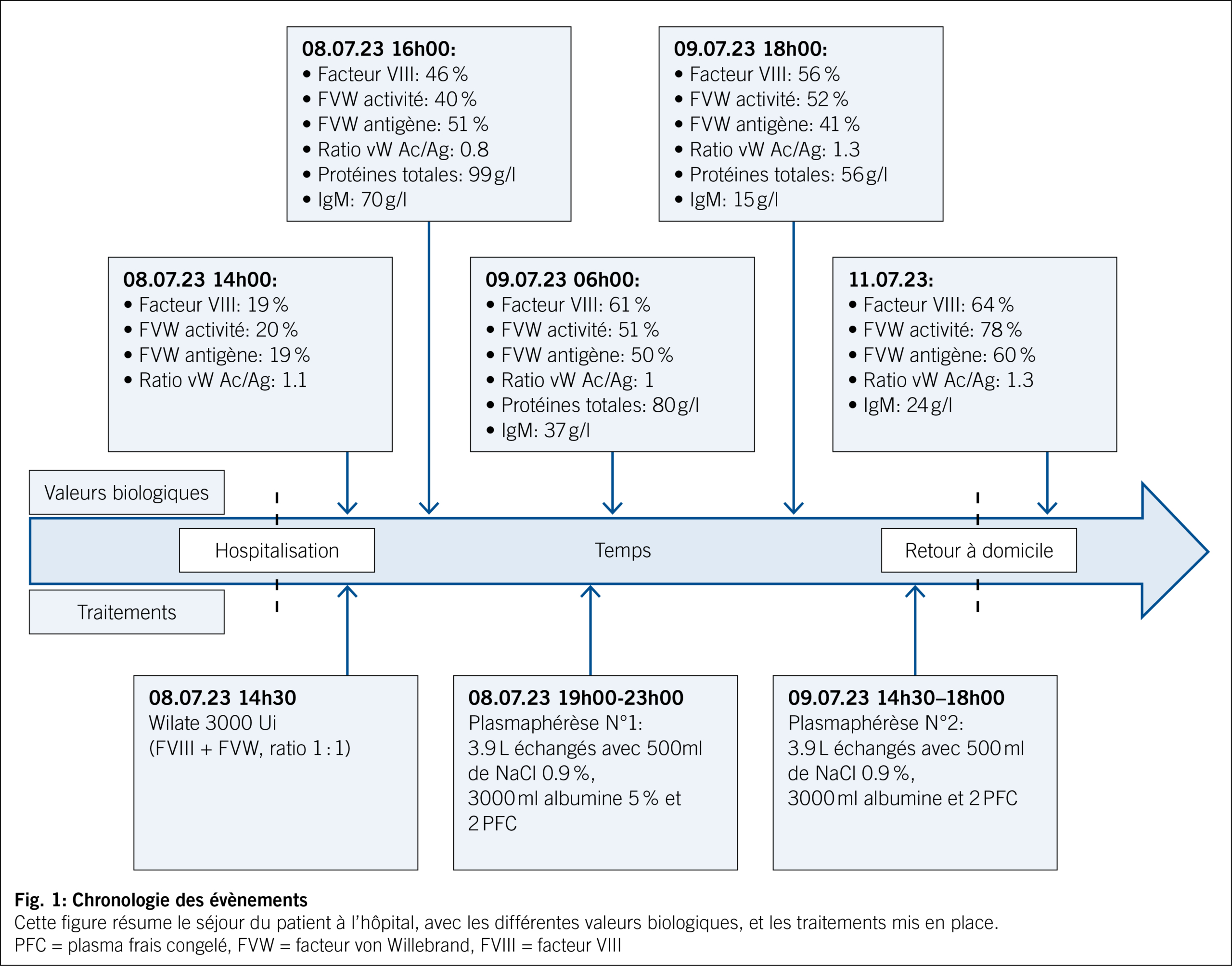
Chronologie des événements
Après la première plasmaphérèse les protéines totales diminuent de 99 g/l à 79 g/l, puis à 56 g/l après la seconde plasmaphérèse. Les deux séances de plasmaphérèse conduisent également à une rapide réduction des IgM (de 70 g/l à 15 g/l) en raison de la distribution majoritairement intravasculaire des IgM (80 % intravasculaire) avec peu de phénomène de rebond après les traitements. Les tests de la crase objectivent une augmentation du FVIII et FVW (Fig. 1). Le patient peut ainsi regagner son domicile, avec poursuite du traitement hémato-oncologique en ambulatoire.
Revue de la litterature
Facteur von Willebrand (VWF)
Le facteur Von Willebrand (FVW) est une grande glycoprotéine multimérique qui joue un rôle crucial dans le système de coagulation du sang. Il doit son nom au médecin finlandais Erik von Willebrand (1870–1949), qui a été le premier à identifier la pathologie liée à son déficit en 1926, en la différenciant de l’ hémophilie (2, 3).
Le FVW est une protéine complexe dont la structure comprend plusieurs domaines fonctionnels. Il existe dans le sang sous différentes tailles (multimères), allant de formes plus petites à des formes plus grandes, de plus en plus adhésives. La synthèse du FVW a lieu dans les cellules endothéliales et les mégacaryocytes. Il est ensuite libéré dans la circulation sanguine, où il circule jusqu’ à ce qu’ il soit nécessaire à l’ hémostase (4, 5).
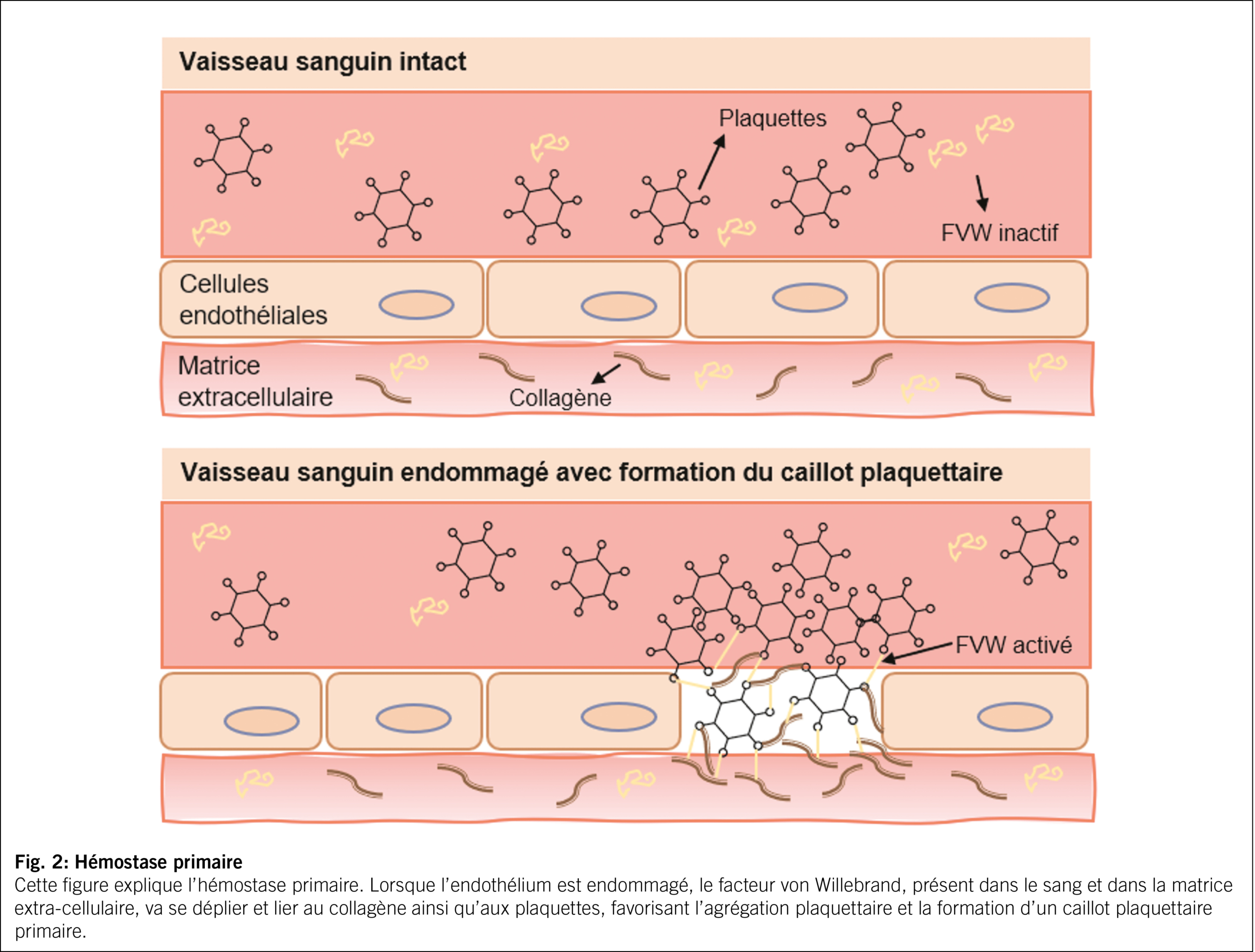
L’ une des deux fonctions du FVW est de faciliter l’ adhésion des plaquettes au site de la lésion vasculaire (Fig. 2). Lorsque les vaisseaux sanguins sont endommagés, exposant le collagène sous-jacent, le FVW se lie au collagène et forme un pont entre la paroi du vaisseau endommagé et les plaquettes. Il joue ainsi un rôle clé dans la formation du clou plaquettaire initial sur le site d’ une lésion vasculaire, favorisant l’ adhésion et l’ agrégation des plaquettes. L’ adhésivité du FVW est directement proportionnelle à la longueur des multimères. Le FVW sert aussi de protéine porteuse pour le facteur VIII, un facteur de coagulation important dans l’ hémostase secondaire. Lorsque le FVW se lie au facteur VIII, il le protège d’ une dégradation rapide, prolongeant ainsi sa présence dans la circulation sanguine et renforçant son activité de coagulation. Le FVW, en enrichissant le FVIII au site de lésion vasculaire, contribue également à la production de la fibrine, le tissu protéique qui renforce le bouchon plaquettaire et forme un caillot sanguin plus durable (6). Un déficit en FVW va donc perturber l’ hémostase primaire et secondaire.
Le déficit héréditaire en facteur von Willebrand entraîne un trouble de la coagulation connu sous le nom de maladie de von Willebrand (MVW). La clinique est souvent peu bruyante. On observe des saignements des muqueuses (gencives et épistaxis), des ecchymoses spontanées, des saignement prolongés ou des saignements menstruels abondants. Il existe différents types de MVW congénitale liée à des anomalies quantitatives (dites de type 1 si le déficit est partiel; type 3 si total) ou qualitatives (types 2) du FWV. La forme acquise, nommée syndrome de von Willebrand, est secondaire à plusieurs pathologies sous-jacentes. Dans cette revue de littérature, nous nous focaliserons sur la forme acquise (2, 7).
Dans le syndrome de von Willebrand acquis (SWa), les niveaux ou la fonction du FVW sont compromis en raison d’ une autre affection médicale. Par exemple, des maladies comme les maladies auto-immunes ou certaines tumeurs malignes peuvent entraîner la production d’ anticorps qui ciblent et détruisent le FVW, contribuant ainsi aux tendances hémorragiques.
Déficit acquis en FVW
a. Rappel épidémiologique
Décrit pour la première fois en 1968 dans le contexte d’ un lupus systémique, le SWa est une pathologie rare, dont la prévalence est certainement sous-estimée en raison du fait que ce diagnostic n’ est que peu souvent évoqué, probablement à cause de la grande variabilité des présentations cliniques. Contrairement à la maladie de von Willebrand congénitale, le SWa se manifeste à un âge plus avancé. Typiquement, on ne retrouve pas d’ antécédents familiaux de diathèse hémorragique. La répartition entre hommes et femmes est égale (8, 9).
b. Physiopathologie
Le syndrome de von Willebrand acquis (SWa) est un trouble rare de la coagulation caractérisé par une déficit acquis quantitatif partiel (type 1) ou qualitatif (type 2) du FVW, secondaire à une pathologie sous-jacente. Le SWa est donc une manifestation d’ une autre affection médicale.
L’ étiologie la plus fréquente est la production par le système immunitaire d’ auto-anticorps qui ciblent le FVW. Ces anticorps entraînent une élimination rapide du FVW de la circulation sanguine, réduisant ainsi son taux et altérant sa fonction. Les anticorps peuvent aussi interférer avec la liaison du FVW aux plaquettes ou faciliter sa dégradation, contribuant ainsi à un déficit en FVW fonctionnel.
Dans le contexte de la macroglobulinémie de Waldenström, les anticorps IgM produits par des lymphocytes anormaux peuvent former des complexes immuns avec le FVW. Ces complexes immuns contribuent à la clairance et à la dégradation du FVW, ce qui entraîne une réduction des niveaux et une déficience fonctionnelle.
D’ autres pathologies peuvent entraîner une protéolyse ou une dégradation accrue du FVW. Des enzymes ou des facteurs activés dans le contexte de certaines maladies peuvent cliver les molécules de FVW, les rendant non fonctionnelles.
Dans certains cas, le SWa peut résulter d’ une diminution de la synthèse du FVW par les cellules endothéliales ou les mégacaryocytes, soit dans le cadre de maladies inflammatoires chroniques qui entraînent un stress oxydatif sur les cellules endothéliales, soit par toxicité de certains médicaments. Dans des autres situations, le FVW est absorbé sur la surface de cellules tumorales.
Les conditions cardiaques qui provoquent une augmentation du stress mécanique ou des turbulences dans les vaisseaux sanguins peuvent contribuer au dépliage et à la dégradation des multimères du FVW par la protéine ADAMTS-13. Ce dépliage réduit l’ activité fonctionnelle du VWF, compromettant sa capacité à participer à la formation de caillots sanguins (10, 11).
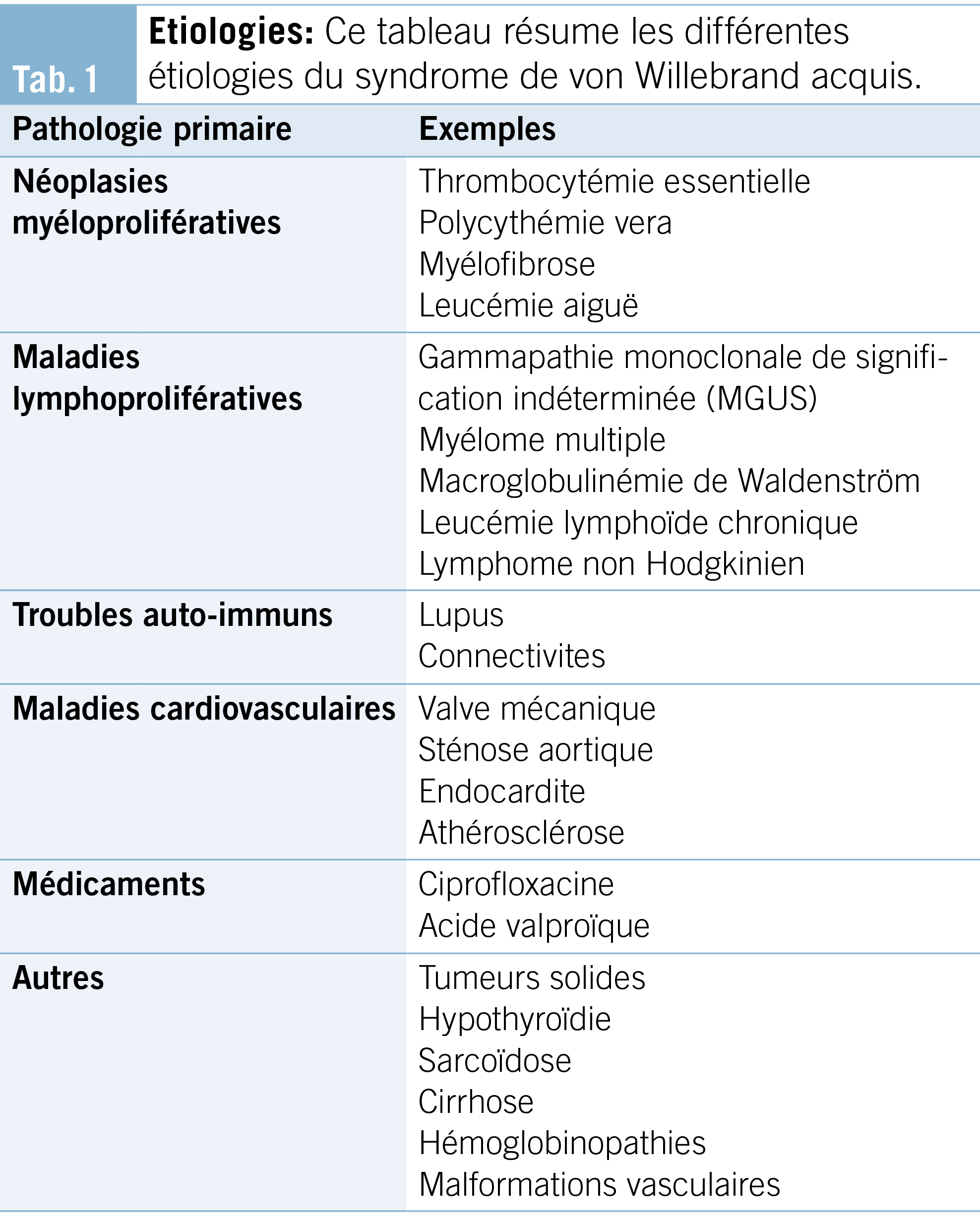
c. Étiologies (Tab. 1)
Le SWa est associé principalement aux maladies myéloprolifératives (thrombocytémie essentielle, polycythemia vera) et lymphoprolifératives (myélome multiple, macroglobulinémie de Waldenström), aux maladies auto-immunes (lupus, connectivites), et aux maladies cardiovasculaires (sténose aortique, valve mécanique). Certains médicaments ont été associés au développement du SWa, comme par exemple, l’ acide valproïque et la ciprofloxacine. Enfin d’ autres causes plus rares comme l’ hypothyroïdie, ou des infections (virales et parasitaires) ont également été décrites comme étiologies (8, 10–13). La gammapathie monoclonale de signification indéterminée (MUGS) et le myélome multiple sont les étiologies les plus fréquemment retrouvées (14).
Le SWa peut se manifester sous forme d’ un défaut quantitatif (type 1) ou qualitatif (type 2), en fonction du mécanisme sous-jacent (diminution de la production, augmentation de la clairance, augmentation de la protéolyse, augmentation de l’ absorption). Par exemple, dans le cas de l’ hypothyroïdie, le SWa est généralement de type 1, suggérant une production réduite de FVW. En revanche, les patients présentant des auto-anticorps, une sténose aortique, ou une protéolyse accrue du FVW peuvent présenter un trouble de type 2 (15).
Dans un registre de la Société internationale de thrombose et d’ hémostase (ISTH) (9) qui a rassemblé les données de 211 cas de SWa, les troubles lymphoprolifératifs (y compris les MGUS et le myélome multiple) étaient la maladie sous-jacente la plus fréquente dans 48 % des cas, tandis que les néoplasies myéloprolifératives et les tumeurs solides représentaient 15 % et 5 % des cas, respectivement. Ainsi, 2/3 des cas de SWa sont liés aux maladies hémato-oncologiques.
Dans deux études de 2015, Mital et al. (16, 17) ont objectivés que sur 312 patients atteints de thrombocytémie essentielle (n=170) ou de polycythemia vera (n=142) la prévalence respective du SWa était de 20 % et 12 %.
À noter que le SWa dans les maladies cardiovasculaires n’ a été étudié que très récemment, et que leur proportion est très probablement sous-estimée (18).
d. Manifestations cliniques
La sévérité de la présentation clinique du SWa peut varier considérablement. Certains cas sont asymptomatiques. Les manifestations cliniques typiques sont des saignements cutanéo-muqueux, des pétéchies, des ecchymoses, des saignements gingivaux, des épistaxis, des saignements menstruels abondants et des saignements gastro-intestinaux. La gravité des symptômes peut varier, et les saignements peuvent être spontanés ou excessifs à la suite d’ interventions chirurgicales ou de blessures. La présentation clinique est ainsi très variable, et peut passer inaperçue pendant de nombreuses années (19).
e. Diagnostic
Le diagnostic du SWa se base sur différents tests de laboratoire. Pour identifier un défaut de l’ hémostase primaire, on peut réaliser un test PFA® (Platelet Function Analyzer) qui a une sensibilité de plus de 90 % pour diagnostiquer un déficit en FVW (20). À ce propos, il faut noter que l’ analyse PFA dépend aussi du taux plaquettaire et de l’ hématocrite et que, malgré sa dénomination, il ne permet pas un screening fiable des fonctions plaquettaires. Pour tester l’ hémostase secondaire, on réalise un aPTT qui peut être prolongé car le facteur VIII est lié au FWV.
Plusieurs tests spécifiques évaluent à la fois le niveau et la fonction du FVW pour déceler une diminution du FVW et/ou de son activité fonctionnelle. Les tests clés traditionnels incluent l’ antigène du FVW (VWF:Ag) et la liaison du FVW à son récepteur plaquettaire (glycoprotéine Ib, GPIb), classiquement mesuré à l’ aide du test du cofacteur de la ristocétine (VWF:RCo) et nouvellement à l’ aide de billes couplées avec un récepteur GPIb hyperfonctionnel (VWF:Ac). Il est également essentiel d’ évaluer l’ activité coagulante du facteur VIII (FVIII:C). D’ autres tests fonctionnels, tels que la liaison du FVW au collagène (VWF:CB), doivent également être pris en compte dans des situations complexes. En général, l’ antigène du FVW (VWF:Ag) sera normal ou légèrement diminué, alors que son activité sera-t-elle nettement diminuée (VWF:Ac). Ainsi le ratio VWF:Ac/VWF:Ag est souvent diminué (< 0.7) (21). Il est nécessaire également de réaliser une électrophorèse pour démontrer le déficit des plus grands multimères de FVW. Enfin, il est également possible de mesurer le propeptide du FVW qui permet de distinguer un défaut de production d’ une clearance augmentée (22).
Il existe ainsi plusieurs tests de laboratoire différents dont aucun n’ est très spécifique. Il faut ainsi analyser les multiples tests réalisés, étudier la chronologie des symptômes (notamment l’ âge auquel les premiers symptômes sont apparus), et mener une anamnèse familiale précise afin de préciser au mieux le diagnostic. Il est important de différencier une maladie de von Willebrand congénitale, d’ une forme acquise, car le traitement varie considérablement. Cette différentiation est facilitée par l’ observation de la cinétique de recirculation (estimation de la demi-vie) du FVW transfusé.
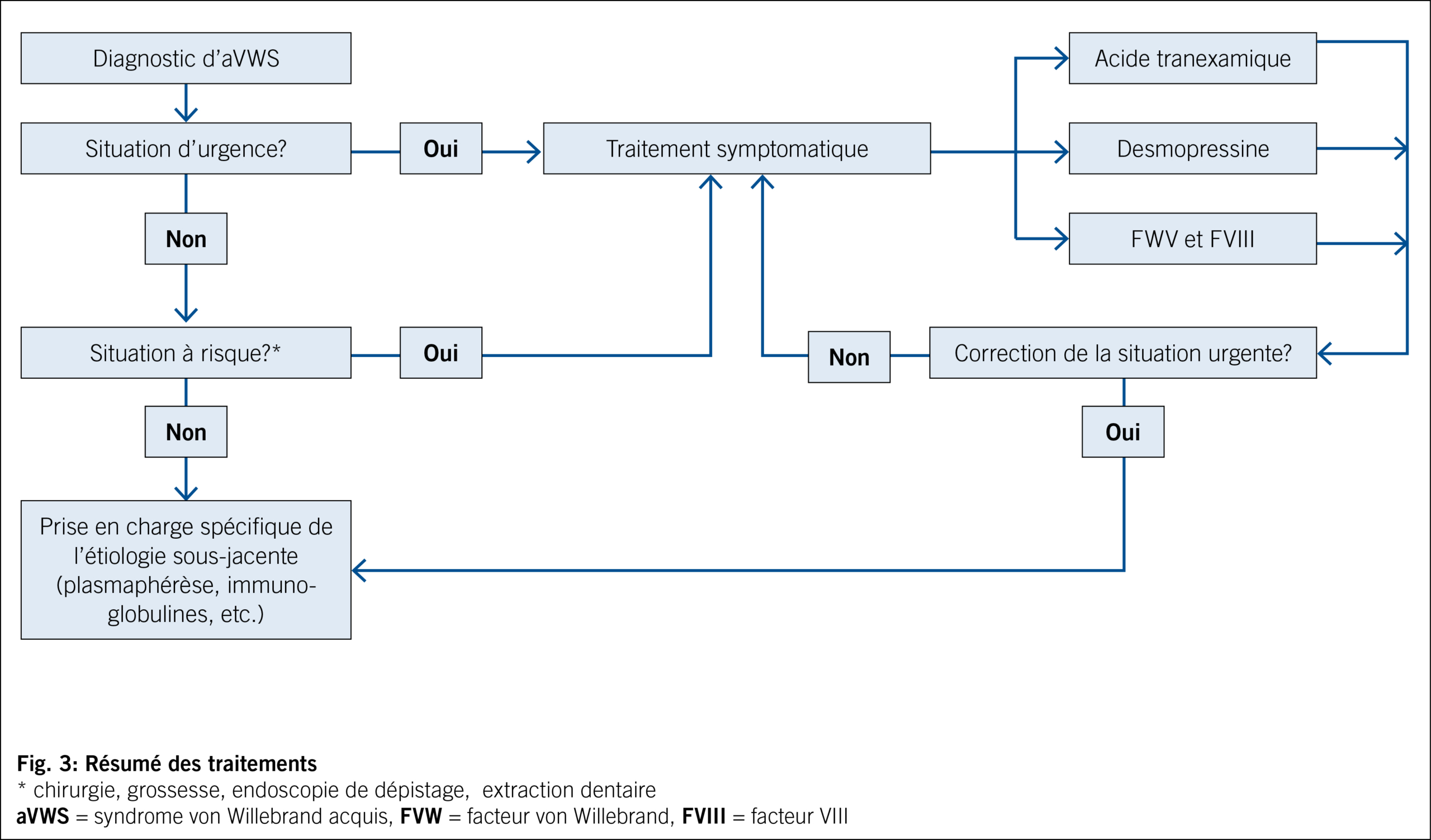
f. Traitement (Fig. 3)
Il existe deux types d’ approche thérapeutique dans le SWa. La première approche consiste à s’ attaquer à la pathologie sous-jacente. Le traitement de la cause associée au SWa conduit à l’ amélioration ou à la résolution du syndrome.
La seconde approche consiste à un traitement symptomatique, soit en prévention d’ une situation à risque (chirurgie, grossesse, colonoscopie de dépistage), soit dans le traitement d’ une situation aiguë (hémorragie). La desmopressine (DDAVP), en stimulant le relargage de FVW des cellules endothéliales, et augmentant le taux plasmatique de FVIII, peut permettre de diminuer le risque hémorragique. Le registre de l’ ISTH a rapporté un taux de réussite global avec le DDAVP d’ environ 30 %. La réponse au traitement est toutefois de brève durée et très variée en fonction de l’ étiologie sous-jacente au SWa: 10 % dans les maladies cardiovasculaires, 21 % dans les néoplasies myéloprolifératives, 33% dans les maladies auto-immunes et 44 % dans les troubles lymphoprolifératifs (8, 12, 13, 23, 24).
La substitution plasmatique en FVW et FVIII a également été étudiée avec des résultats favorables. Environ 40 % des patients souffrant de SWa présentent une bonne réponse. La substitution nécessite toutefois un monitoring biologique continu car les réponses sont très variables en termes de durée (8, 12, 13, 25). Les traitements fibrinolytiques, tel l’ acide tranexamique (Cyklokapron®), peuvent également jouer en rôle dans les situations d’ urgence (26).
Les immunoglobulines intraveineuses (IVIG) ont démontré un bon effet dans certains cas, notamment les SWa associés aux tumeurs solides, ainsi que dans les processus auto-immuns et lymphoprolifératifs à IgG (MGUS, myélome multiple) (12, 13, 25). Enfin la plasmaphérèse, qui va éliminer les auto-anticorps et les paraprotéines, est utilisée principalement dans les maladies à IgM.
Maladie de Waldenström
La macroglobulinémie de Waldenström (MW) est un type rare de lymphome non hodgkinien caractérisé par la prolifération de globules blancs anormaux (lymphocytes B) dans la moelle osseuse et la surproduction d’ un anticorps spécifique IgM. L’ âge de survenue moyen est de 71 ans. L’ incidence est faible: 0.3 cas par 100 000 habitants par année. Les symptômes courants sont la faiblesse, la fatigue, l’ anémie, l’ hypertrophie des ganglions lymphatiques, les atteintes neurologiques (23 % de polyneuropathies au diagnostic), et, dans certains cas, le syndrome d’ hyperviscosité dû à des taux élevés d’ IgM dans le sang. Le risque hémorragique est également augmenté, en lien avec plusieurs complications possibles de la MW: SWa, amyloïdose, cryoglubulinémie, hématopoïèse inefficace. Le diagnostic se pose par immunofixation et biopsie de moelle osseuse (27–29).
Dans le contexte de la macroglobulinémie de Waldenström, les anticorps IgM produits par des lymphocytes anormaux peuvent former des complexes immuns avec le FVW. Ces complexes immuns contribuent à la clairance et à la dégradation du FVW, ce qui entraîne une diminution de son taux et une déficience fonctionnelle. Le SWa survient chez 6% des patients atteints de MW, et son incidence augmente proportionnellement au taux d’ IgM. Plus le taux d’ IgM est élevé, plus le risque d’ un SWa est important (30).
Le traitement se base ainsi sur l’ élimination des IgM, pour en diminuer le taux dans des valeurs en dessous de 30 à 60 g/L. La plasmaphérèse est le traitement de choix, mais implique la pose de voies veineuses de gros calibre, avec un risque hémorragique important. Un traitement adjuvant par desmopressine et concentrés de FVW/FVIII est par conséquent utilisé (28, 30, 31). La diminution du taux d’ IgM permet en général de traitement du syndrome von Willebrand acquis.
Conclusion
Le SWa est une pathologie rare, complexe et variée. Sa prévalence est fortement sous-estimée. De nombreuses conditions médicales sont liées à ce syndrome, notamment les hémopathies malignes, les pathologies auto-immunes et certaines pathologies cardiovasculaires. Le diagnostic est posé sur la base d’ une clinique évocatrice, de l’ absence d’ anamnèse familiale pour une diathèse hémorragique, et de divers tests de laboratoire. Le défi réside ensuite dans l’ identification de la pathologie sous-jacente. Son identification est primordiale car elle va guider le traitement causal. Des traitements de support, comme les concentrés plasmatiques en FVW et FVIII, ainsi que les antifibrinolytiques sont utilisés notamment dans les situations d’ urgence, et en prévention de complications dans des situations à risque, comme les chirurgies.
Service de médecine interne
Centre Hospitalier Universitaire Vaudois
Rue du Bugnon 46,
CH-1011 Lausanne, Suisse
vincent.jendly@chuv.ch
Service de néphrologie,
Centre Hospitalier Universitaire Vaudois (CHUV) and Université de Lausanne (UNIL),
Lausanne
FMH médecine interne et hématologie FMH/FAMH,
Centre d’ hématologie CMCV,
La Chaux de Fonds
FMH médecine interne et hématologie FMH/FAMH,
Centre d’ hématologie CMCV,
La Chaux de Fonds
Service de néphrologie,
Centre Hospitalier Universitaire Vaudois (CHUV) and Université de Lausanne (UNIL),
Lausanne
Service et Laboratoire central d’ hématologie,
Centre Hospitalier Universitaire Vaudois (CHUV) and Université de Lausanne (UNIL),
Lausanne
Les auteurs n’ ont pas déclaré de conflit d’ intérêts en rapport avec cet article
1. Connelly-Smith L, Alquist CR, Aqui NA, Hofmann JC, Klingel R, Onwuemene OA, Patriquin CJ, Pham HP, Sanchez AP, Schneiderman J, Witt V, Zantek ND, Dunbar NM. Guidelines on the Use of Therapeutic Apheresis in Clinical Practice – Evidence-Based Approach from the Writing Committee of the American Society for Apheresis: The Ninth Special Issue. J Clin Apher. 2023 Apr;38(2):77-278. doi: 10.1002/jca.22043. PMID: 37017433.
2. F. Boehlen, H. Robert-Ebadi, P. de Moerloose, Rev Med Suisse 2007 ; 3 : 346-50
3. Fontana, P., Boehlen, F., Hémostase diagnostic et prise en charge des syndromes hémorragiques dits mineurs, Rev Med Suisse, 2008/140 (Vol.-6), p. 122–126.
4. Sadler JE, Budde U, Eikenboom JC, Update on the pathophysiology and classification of vonWillebrand disease: A report of the Subcommittee on von Willebrand Factor. J Thromb Haemost 2006; 4:2103–14
5. Fressinaud E, Meyer D. La maladie de vonWillebrand: du diagnostic au traitement. Rev Prat 2005;55: 2209–18
6. Cortes GA, Moore MJ, El-Nakeep S. Physiology, Von Willebrand Factor. 2023 Feb 20. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan–. PMID: 32644488
7. TosettoA, Rodeghiero F, Castaman G, A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: Results from a multicenter European study (MCMDM-1 VWD). J Thromb Haemost 2006;4:766–73
8. Franchini M, Mannucci PM. Acquired von Willebrand syndrome: focused for hematologists. Haematologica. 2020 Aug;105(8):2032-2037.. Epub 2020 Jun 18. PMID: 32554559; PMCID: PMC7395262.
9. Federici AB, Budde U, Rand JH. Acquired von Willebrand syndrome 2004: International Registry–diagnosis and management from online to bedside. Hämostaseologie. 2004 Feb;24(1):50-5. PMID: 15029273.
10. Thomas J, Kostousov V, Teruya J. Bleeding 30. and thrombotic complications in the use of extracorporeal membrane oxygenation. Semin Thromb Hemost. 2018;44(1):20-29
11. Nascimbene A, Neelamegham S, Frazier OH, Moake JL, Dong JF. Acquired von Willebrand syndrome associated with left ventricular assist device. Blood. 2016 Jun 23;127(25):3133-41. doi: 10.1182/blood-2015-10-636480. Epub 2016 May 3. PMID: 27143258; PMCID: PMC4920020.
12. Federici AB, Rand JH, Bucciarelli P, et al. 26. Acquired von Willebrand syndrome: data from an International registry. Thromb Haemost. 2000;84(2):345-349.
13. Federici AB, Budde U, Castaman G, Rand JH, Tiede A. Current diagnostic and therapeutic approaches to patients with acquired von Willebrand syndrome: a 2013 update. Semin Thromb Hemost. 2013;39(2):191-201
14. Mital A. Acquired von Willebrand Syndrome. Adv Clin Exp Med. 2016 Nov-Dec;25(6):1337-1344. doi: 10.17219/acem/64942. PMID: 28028990.
15. Franchini M, Lippi G, Favaloro EJ. Advances in hematology. Etiology and diagnosis of acquired von Willebrand syndrome. Clin Adv Hematol Oncol. 2010 Jan;8(1):20-4. PMID: 20351678.
16. Mital A, Prejzner W, Bieniaszewska M, Hellmann A. Prevalence of acquired von Willebrand syndrome during essential thrombocythemia: a retrospective analysis of 170 consecutive patients. Pol Arch Med Wewn. 2015;125(12):914-920.
17. Mital A, Prejzner W, Swia ̨tkowska Stodulska R, Hellmann A. Factors predisposing to acquired von Willebrand syndrome during the course of polycythemia vera – ret- rospective analysis of 142 consecutive cases. Thromb Res. 2015;136(4):754-757.
18. Vincentelli A, Susen S, Le Tourneau T, Six I, Fabre O, Juthier F, Bauters A, Decoene C, Goudemand J, Prat A, Jude B: Acquired von Willebrand syndrome in aortic stenosis. N Engl J Med 2003, 349, 343–349
19. Langer AL, Connell NT. Acquired von Willebrand Syndrome. Hematol Oncol Clin North Am. 2021 Dec;35(6):1103-1116. doi: 10.1016/j.hoc.2021.07.005. Epub 2021 Aug 12. PMID: 34391604.
20. Reber G, Boehlen F, Fontana P. Investigation de l’ hémostase primaire in vitro à l’ aide du Platelet Function Analyzer (PFA-100TM). Rev Med Suisse 2003;123:491-4
21. Favaloro EJ, Facey D, Grispo L. Laboratory assessment of von Willebrand factor. Use of different assays can influence the diagnosis of von Willebrand’ s disease, dependent on differing sensitivity of sample preparation and differential recognition of high molecular weight VWF forms. Am J Clin Pathol. 1995;104(3):264-271
22. Eikenboom J, Federici AB, Dirven RJ, et al; MCMDM-1VWD Study Group. VWF propeptide and ratios between VWF, VWF propeptide, and FVIII in the characterization of type 1 von Willebrand disease. Blood.2013;121(12):2336-2339
23. Franchini M. The use of desmopressin as a 619. hemostatic agent: a concise review. Am J Hematol. 2007;82(8):731-735.
24. Biguzzi E, Siboni SM, Peyvandi F. Acquired von Willebrand syndrome and response to desmopressin. Haemophilia. 2018;24(1):e25- e28
25. Federici AB, Budde U, Rand JH. Acquired von Willebrand syndrome 2004: internation- 27. al registry. Hämostaseologie. 2004;24(1):50-55.
26. Franchini M, Mannucci PM. The never ending success story of tranexamic acid in acquired bleeding. Haematologica. 2020;105 (5):1201-1205
27. Baďurová K, Gregorová J, Vlachová M, Krejčí M, Ševčíková S. Waldenström macroglobulinemia. Klin Onkol. 2021 Fall;34(6):428-433. English. doi: 10.48095/ccko2021428. PMID: 34911327.
28. Brysland SA, Maqbool MG, Talaulikar D, Gardiner EE. Bleeding Propensity in Waldenström Macroglobulinemia: Potential Causes and Evaluation. Thromb Haemost. 2022 Nov;122(11):1843-1857. doi: 10.1055/a-1896-7092. Epub 2022 Jul 11. PMID: 35817084; PMCID: PMC9626029.
29. Talaulikar D, Tam CS, Joshua D, et al. Treatment of patients with Waldenström macroglobulinaemia: clinical practice guidelines from the Myeloma Foundation of Australia Medical and Scien- tific Advisory Group. Intern Med J 2017;47(01):35–49
30. Castillo JJ, Gustine JN, Meid K, et al. Low levels of von Willebrand markers associate with high serum IgM levels and improve with response to therapy, in patients with Waldenström macroglo- bulinaemia. Br J Haematol 2019;184(06):1011–1014
31. Abou-Ismail MY, Rodgers GM, Bray PF, Lim MY. Acquired von Willebrand syndrome in monoclonal gammopathy – a scoping review on hemostatic management. Res Pract Thromb Haemost 2021;5(02):356–365
PRAXIS
- Vol. 113
- Ausgabe 8
- September 2024