Die thrombotische Mikroangiopathie (TMA) wird durch die typische Trias einer schweren Thrombozytopenie, Coombs-negativen hämolytischen Anämie sowie Endorgandysfunktion definiert. Pathophysiologisch handelt es sich um Ischämie-bedingende Mikrothromben in Arteriolen und Kapillaren, welche zu schwerwiegender Organdysfunktion sowie akut lebensbedrohlichen Endorganschädigungen führen können. Hinsichtlich Ätiologie, Verlauf, Therapie und Prognose werden die folgenden Manifestationsformen unterschieden: die thrombotisch-thrombozytopenische Purpura (TTP), das Shigatoxin-induzierte hämolytisch-urämische Syndrom (STEC-HUS), die sekundäre TMA und das atypische hämolytisch-urämische Syndrom (aHUS).
Wir präsentieren den Fall eines 49-jährigen lungentransplantierten Patienten mit aHUS. Die Komplexität der zugrunde liegenden Pathomechanismen der TMA, die schwierige Differenzierung der TMA-Manifestationen und das anspruchsvolle Management eines aHUS nach Lungentransplantation verdeutlichen die Einzigartigkeit dieses Patientenfalles.
Schlüsselwörter: thrombotische Mikroangiopathie, atypisches hämolytisch-urämisches Syndrom, Eculizumab, Lungentransplantation
Thrombotische Mikroangiopathie: Manifestationsformen
Die TTP resultiert aus einer genetisch bedingten oder erworbenen Reduktion der Aktivität der von-Willebrand-Faktor (vWF) spaltenden Metalloproteinase ADAMTS13. Bei der selteneren kongenitalen TTP kommt es mutationsbedingt zu einer verringerten hepatischen Produktion von ADAMTS13. Die deutlich häufigere erworbene Form, auch «acquired TTP» (aTTP) genannt, betrifft vor allem junge Erwachsene ohne Vorerkrankungen und ist in einer IgG-Autoantikörper vermittelten Funktionseinschränkung des Enzyms begründet (1). Die aTTP, welche ätiologisch die Mehrheit aller TTP-Fälle ausmacht, gehört zur Gruppe seltener Erkrankungen mit einer jährlichen Inzidenz von 1.5 bis 6 Fällen pro 1 Million Einwohner in Europa (2, 3). Der Defekt der Protease resultiert in einer Multimerbildung des vWF-Glykoproteins.
Diese bedingen eine aggravierte Thrombozytenaggregation mit folgender mikrovaskulärer Thrombosierung und Hämolyse (4). Diagnostisch wegweisend ist die klinische Präsentation einer renalen Dysfunktion mit begleitenden neurologischen Symptomen in Kombination mit Thrombozytopenie und hämolytischer Anämie (4, 5). Im Falle der sekundären TMA ist die Ätiologie vielseitig; beschriebene Auslöser reichen von bakteriellen, viralen und fungale Infektionen, Krebserkrankungen, Autoimmunerkrankungen (z. B. systemischer Lupus Erythematodes, systemische Sklerose, Antiphospholipid- Antikörper-Syndrom), Organ- oder Knochenmarktransplantationen und Schwangerschaft bis zu Drogenkonsum sowie medikamentösen Triggern (4). Fall-Kontroll-Studien aus dem angloamerikanischem Raum beschreiben maligne Erkrankungen als die häufigsten Risikofaktoren einer sekundären TMA (2). Im Falle einer nicht ADAMTS13-Aktivität bedingten TMA führt eine systemische Endothelschädigung durch immunzelluläre und Komplement-abhängige Pathomechanismen zur mikrovaskulären Thrombosierung.
Das STEC-HUS, auch typisches HUS genannt, ist eine Komplikation im Rahmen einer gastrointestinalen Infektion mit Shigatoxin-bildenden Erregern – meist enterohämorrhagischen Escherichia coli. Das STEC-HUS tritt typischerweise bei Kindern im Alter zwischen zwei und fünf Jahren auf, kann allerdings im Rahmen von epidemischen Ausbrüchen auch andere Altersgruppen betreffen (6). Die Zerstörung renaler und intestinaler Endothelzellen ergibt das klinische Bild von meist blutigen Durchfällen und akuter Nierenfunktionseinschränkung (AKI).
Im Falle des atypischen HUS (aHUS) besteht eine endogene Prädisposition für ein hyperreagibles Komplementsystem. Zeitpunkt und Schwere der klinischen Manifestation sind höchst individuell und meist mit dem Auftreten endo- beziehungsweise exogener Stressoren assoziiert, sogenannter Komplement-aggravierender Faktoren. Ursächlich ist ein genetisch bedingter Funktionsverlust von Komplementregulatoren, das Vorliegen von Gain-of-function-Mutationen in Komplement-codierenden Genabschnitten oder eine Autoantikörper bedingte Hemmung von Komplementinhibitoren (4). Obwohl das aHUS als primäre Erkrankung des Komplementsystems definiert ist, handelt es sich im klinischen Alltag oft um eine differenzialdiagnostische Ausschlussdiagnose. Das heisst, es gibt keinen laborchemischen Test, welcher die Erkrankung im akuten Setting eindeutig diagnostizieren kann. Sie wird als «ultra-rare-disease» eingestuft, mit einer geschätzten jährlichen Inzidenz weltweit von 0.23–1.9/Million (7, 8, 9). Die Prognose ist mit einer Mortalität von 25 % und einem Patientenanteil von bis zu 50 %, der in der Akutphase ein Nierenversagen entwickelt, deutlich schlechter als beim typischen HUS, welches mit einer Letalität von 1–5 % die beste Prognose aller TMA-Manifestationen besitzt (10–12).
Fallbericht
Zum ersten Mal vorstellig wurde unser Patient im Oktober 2021 mit pulmonal rasch progredienter Symptomatik im Sinne einer connective tissue disease-associated interstitial lung disease (CTD-ILD) bei neu diagnostiziertem Sjögren- Syndrom. Der Patient präsentierte eine rasche lungenfunktionelle Verschlechterung im Sinne einer progredienten restriktiven Ventilations- und Diffusionsstörung, die innerhalb weniger Monate zu einer respiratorischen Insuffizienz führten. Dank frühzeitiger Zuweisung konnte der Patient nach einer zweiwöchigen Lungentransplantationsabklärung im Universitätsspital Zürich im Oktober 2022 bilateral lungentransplantiert werden.
Ende April 2023 stellte sich der Patient mit Thoraxschmerzen, Kopfschmerzen und Verschlechterung des Allgemeinzustands vor. Es präsentierte sich eine Panzytopenie mit aggravierter Thrombozytopenie (Nadir 12 g/l), deutlich erhöhter LDH, erhöhtem Bilirubin, einer transfusionspflichtigen hämolytischen Anämie (Hb 51 g/l) und rapider Verschlechterung der Nierenfunktion im Rahmen einer akuten Nierenfunktionseinschränkung Stufe 3 nach KDIGO. Die mikroskopische Zelldifferenzierung zeigt 20–25 Fragmentozyten pro Gesichtsfeld im mikroskopisch hypochrom-anisozytären Blutbild. Der direkte Antiglobulintest (Coombs-Test) zeigte sich negativ (DAT-). Die Parameter Folsäure, Vitamin B12 und Ferritin befanden sich im Normbereich. Eine Neutropenie war bekannt und wurde im Rahmen der knochenmarksuppressiven Medikation des lungentransplantierten Patienten (Ganciclovir, Itraconazol, Mycophenolat mofetil [MMF]) interpretiert. Zum Zeitpunkt der Vorstellung war der Patient aufgrund erhöhter Donor-spezifischen Antikörpern im Serum mit Tacrolimus, MMF, dem mTOR-Inhibitor Everolimus und Prednison vierfach immunsupprimiert.
In der Zusammenschau konnte bei obig bereits beschriebener Thrombozytopenie, transfusionspflichtiger hämolytischer Anämie und rapider Verschlechterung der Nierenfunktion noch am Vorstellungstag die klinische Verdachtsdiagnose einer TMA gestellt werden. Bei annähernd normwertiger ADAMTS13-Aktivität (45 %) sowie negativer EHEC-/Shigatoxin-Testung im Stuhl konnten eine TTP sowie ein STEC-HUS schnell ausgeschlossen werden (Abb. 1).

Es ergaben sich die Differenzialdiagnosen einer sekundären medikamentösen TMA, am ehesten unter kombinierter Calcineurin- und mTOR-Inhibitor-Therapie oder eines atypischen HUS. Um eine mögliche medikamentöse Ursache zu adressieren, wurde ein Calcineurin-Inhibitor-Wechsel innerhalb der Substanzklasse (Tacrolimus zu Ciclosporin A) vorgenommen sowie Everolimus pausiert. Zur weiteren Evaluation einer Komplement-assoziierten TMA im Rahmen eines aHUS wurde die Konzentrationsbestimmung der Komplementfaktoren vorgenommen. Eine Erhöhung von Komplement CD5b-9 MAC konnte bestätigt werden. C3, C4, Faktor H und I waren hingegen unauffällig.
Die vaskulitische Assoziation der hier als Grundmorbidität vorliegende Kollagenose eines Sjögren-Syndroms spielt als autoimmunologischer, prädisponierender Faktor ebenfalls eine Rolle im diagnostischen Abklärungsprozess bei Verdacht einer sekundären TMA (Tab. 1), insbesondere im Falle des hier am ehesten vorliegenden aHUS.

Es erfolgte eine molekulargenetische Diagnostik zur Identifizierung pathogener Mutationen in Komplement-assoziierten Genen (Next Generation Sequencing panel aHUS). Es lag eine Duplikation der im Tandem auf Chromosom 1q lokalisierten mit aHUS assoziierten Gene CD46, CFH und CFHR1–5 vor. Eine Knochenmarkpunktion zeigte lediglich hyporegeneratorische Zellreihen ohne Malignitätshinweise und verblieb somit ohne Hinweis auf ein myeloproliferatives Syndrom.
Ergänzend erfolgte eine Nierenbiopsie, deren Befund mit einem aHUS vereinbar war. Nach interdisziplinärer Rücksprache wurde umgehend mit einer empirischen anti-komplementären Therapie mit Eculizumab begonnen. Unter bestehender Immunsuppression mittels Ciclosporin, MMF und Prednison nach Transplantation wurde unser Patient vor Beginn der Eculizumab-Therapie gegen Meningokokken und Pneumokokken geimpft. Da bei Triple-Immunsuppression nur eine geringe Impfantwort zu erwarten ist, wurde eine antiinfektive Meningokokkenprophylaxe mit Ciprofloxacin 500mg/d begonnen, die bis mindestens sechs Monate nach letzter Eculizumab-Gabe fortgeführt wurde.
Trotz initial raschen Ansprechens – demonstriert durch steigende Thrombozytenwerte, sich stabilisierendem Hämoglobin sowie einer verbesserten Nierenfunktion – musste das Arzneimittel aufgrund ungeklärter finanzieller Kostenübernahme nach lediglich vier Wochen erfolgter Therapie pausiert werden. Nach zwei Wochen Therapiepause kam es zum TMA-Rezidiv mit erneut fallenden Thrombozyten und Erythrozytenzahlen sowie Verschlechterung der Nierenfunktion.
Das klinische Ansprechen auf Eculizumab konnte nach Wiederbeginn der Therapie gänzlich objektiviert werden (Diagnosis ex juvantibus). Es kam erneut zu einer Stabilisierung der Nierenwerte und der Hämatologie. Die rezidivierende Messung der Komplementaktivität und von C5b-9 zeigte eine gute Suppression des Komplementverbrauchs auf dem alternativen Komplementweg unter der Antikörpertherapie.
Diagnostik
Die Verdachtsdiagnose einer TMA wird bei einer absoluten oder relativen Thrombozytopenie (Thrombozyten < 150 G/l, > 25 % Reduktion von der Baseline) in Kombination mit einer Coombs-negativen hämolytischen Anämie mit Fragmentozyten im Blutausstrich, erhöhter LDH, erhöhtem Bilirubin, reduziertem Haptoglobin und Retikulozytose (kann bei Nierenversagen fehlen) sowie Endorgandysfunktion gestellt (Tab. 1). Entscheidend ist nach Diagnose einer TMA, im Anschluss die Ätiologie korrekt zuordnen zu können, um eine zielgerichtete Therapie zu ermöglichen, da sich die Therapie je nach Ätiologie erheblich unterscheidet. Für das optimale Management der mitunter lebensbedrohlichen Organmanifestationen der TMA ist eine kontinuierliche, interdisziplinäre Zusammenarbeit zwischen Nephrologen, Hämatologen, Neurologen und Genetikern essenziell.
Eine frühzeitige Bestimmung der ADAMTS13-Aktivität hat das Ziel, eine TTP als Ursache rasch auszuschliessen. Es folgt die Beurteilung etwaiger infektiöser Ätiologien mit der Durchführung von Real-Time PCR/ELISA zur Testung auf Shigatoxin im Stuhl oder einem Analabstrich sowie einer Röntgen-Thorax-Aufnahme, der Abnahme von Blut-, Sputum, Stuhl- und Urinkulturen, einem Rachenabstrich auf Influenza, Testung auf Pneumokokken-Ag im Urin sowie dem Ausschluss von HIV, Hepatitis B und C. Weiterhin sollten relevante Komorbiditäten und Medikamente als mögliche Auslöser in einer detaillierten Anamnese erfasst werden. Die in Tab. 2 aufgeführten Analysen gelten lediglich als Startpunkt der erweiterten Abklärung und müssen je nach klinischem Verdacht gezielt erweitert werden.
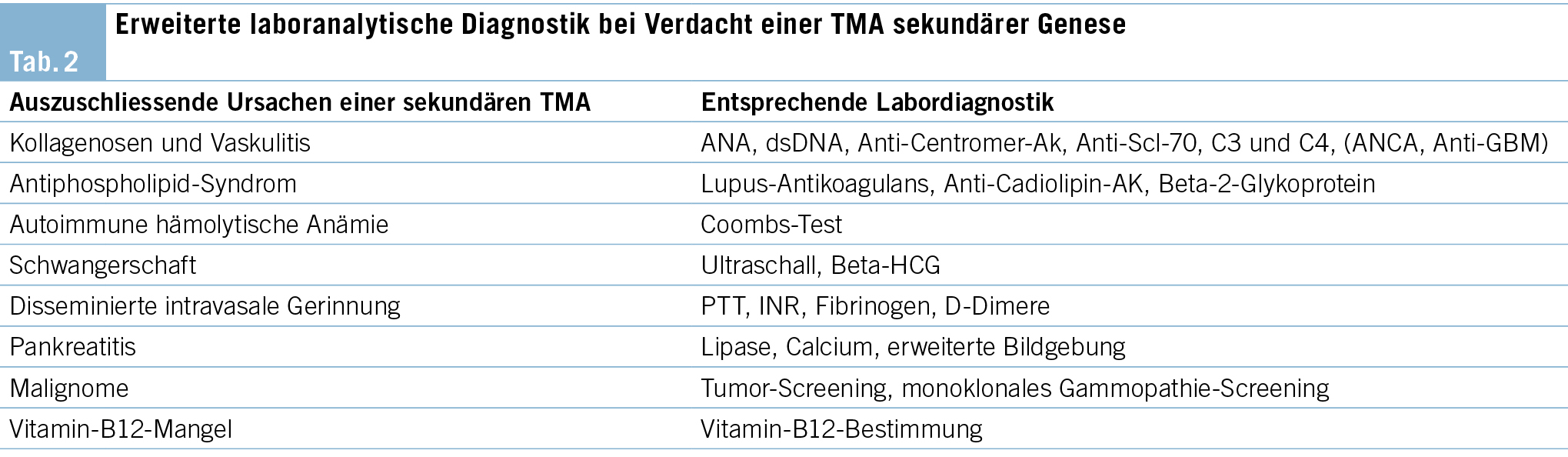
Eine Nierenbiopsie wird nicht nur zur Bestätigung einer TMA als Ursache der Nierenschädigung herangezogen, sondern erlaubt auch die Einschätzung, inwiefern bereits ein chronischer Schaden besteht, und ermöglicht somit eine Aussage über die Prognose. Manchmal kann sie auch Hinweise auf die Ursache der TMA liefern. In der klinischen Praxis wird eine Nierenbiopsie aufgrund der Thrombozytopenie und dem damit einhergehenden Blutungsrisiko initial oft verzögert durchgeführt.
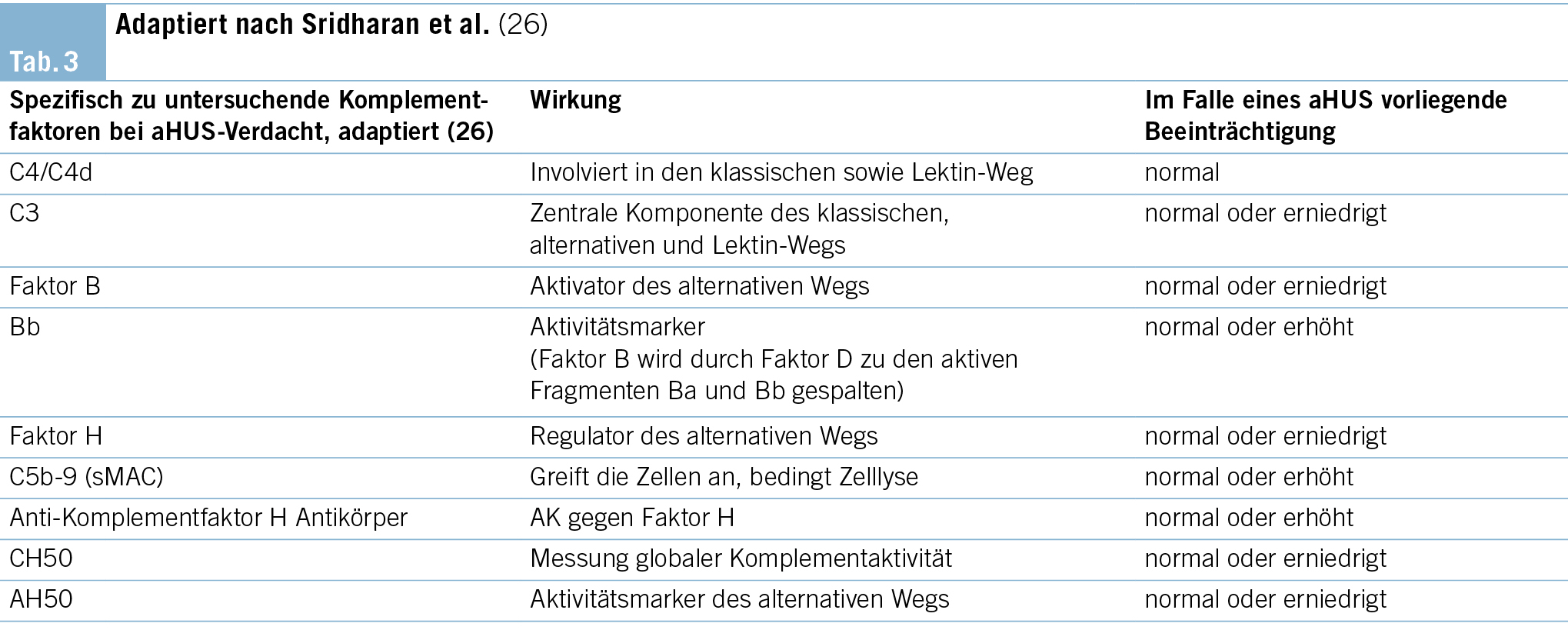
Zeigen die genannten Untersuchungen unauffällige Befunde an, sollte eine Komplementfaktoraktivitätsprüfung kombiniert mit einer genetischen Analyse erfolgen. Bei aHUS-Patienten kann bei ungefähr 40–60 % aller Patienten eine genetische Veränderung im Bereich der Komplementkaskade nachgewiesen werden (13). Tab. 3 führt die wichtigsten Komplementfaktoren, Komplementantikörper und Komplement-codierenden Gene zur Testung auf. Die Bestimmung von Komplementfaktoren und die Sequenzierung von Risikogenen hilft jedoch nicht bei der initialen Diagnosestellung eines aHUS, respektive bei der Therapieentscheidung, ob eine Komplementblockade mit Eculizumab begonnen werden soll, da die Analysen längere Zeit in Anspruch nehmen.
Zum jetzigen Zeitpunkt sind Mutationen in mehr als zehn unterschiedlichen Komplement-codierenden Genen bekannt. Die häufigste und somit klinisch relevanteste Mutation betrifft den hepatisch synthetisierten Komplementfaktor H auf dem CFH-Gen. Weiterhin wichtig und als Hochrisikomutationen bekannt sind Gain-of-function-Mutationen in codierenden Abschnitten von den Komplement-Faktoren B (CFB) und C3 (13).
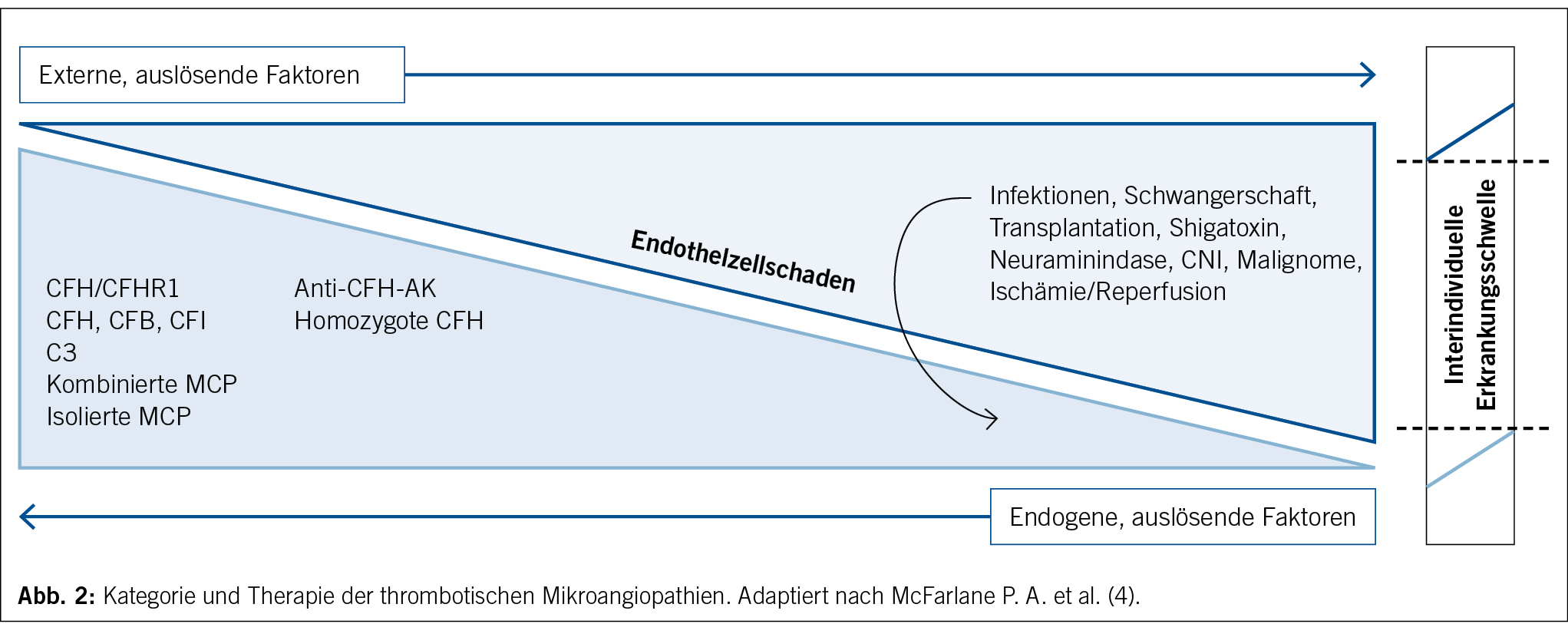
Wie bei vielen anderen hereditären Erkrankungen geht man auch bei der Pathogenese des aHUS von einer Two-Hit-Hypothese aus. Dies bedeutet, dass eine Genmutation allein nicht zwingend die Krankheit zur klinisch-apparenten Manifestation bringt. Studien haben gezeigt, dass selbst bei einer schweren CFH-Mutation die Penetranz bei unter 50 % liegt (13). Komplement-aggravierende Faktoren führen zur Überschreitung der interindividuellen Belastungsschwelle und folglich der Aktivierung des Komplementsystems bei bereits minimalen ersten Endothelschäden (Abb. 2) (13). Bei zugrunde liegender genetischer Prädisposition entwickelt sich daraus eine unkontrollierte, überschiessende systemische Komplementreaktion, welche die fulminante Manifestation der Erkrankung mit folgenden Endorganschäden bedingt (13) (14). Die daraus resultierende Hämolyse aggraviert das überaktive Komplementsystem weiter, sodass ein sich selbst unterhaltender Kreislauf entsteht, welcher den für die Erkrankung so typischen schwerwiegenden klinischen Verlauf bedingt. Weitere Charakteristika eines aHUS, die eine gewisse Abgrenzung zu anderen TMA-Manifestationen erlauben, sind ein familiär gehäuftes Auftreten und ein beobachtetes Rezidiv der Erkrankung nach Transplantation (4).
Therapieansätze
Ursprünglich bestand für Patienten mit TMA als einzige Therapieoption, nebst supportiven Massnahmen, der wiederholte Austausch des Blutplasmas (Plasmapherese). Für Patienten mit aHUS konnte dieser Ansatz selten einen ausreichenden therapeutischen Erfolg erzielen, da die zugrunde liegende Pathogenese nicht behoben wurde. Vor Einführung von komplementinhibierenden Substanzen wurden bis zu 40 % der Patienten trotz Plasmaaustausch oder sonstiger supportiver Therapie bereits mit der ersten klinischen Manifestation dialysebedürftig oder verstarben (10) (15). Im Fünf-Jahres-Verlauf betraf dies bis zu 64 % der adulten Patienten (1) (16).
Während der fulminanten Erstmanifestationsphase bis zum Erhalt der ADAMTS13-Bestimmung ist die Plasmapherese, heutzutage meist unterstützend mit Gabe von Fresh Frozen Plasma (FFP) (17) (Tab. 4), bei adulten Patienten mit TMA unklarer Ätiologie die Therapie der Wahl, da die Verzögerung der Therapie der TTP das klinische Outcome deutlich verschlechtert (4).

Im Falle einer bestätigten TTP besteht eine neue Therapieoption mittels Anti-vWF-Nanobody Caplacizumab. Es reduziert die Adhäsion zwischen grossen vWF-Multimeren und Blutplättchen und führt zu einer rascheren Normalisierung der Thrombozytenzahl, Reduktion der Therapiedauer sowie Wahrscheinlichkeit eines TTP-Rückfalls (17).
Sobald eine TTP ausgeschlossen werden kann und sekundäre Ursachen als unwahrscheinlich erachtet werden, ist mittlerweile die intravenöse Komplementinhibition mittels monoklonalen IgG-Antikörpern, Eculizumab oder Ravulizumab die kausale Therapie der Wahl einer Komplement-vermittelten TMA, da sie zur signifikanten Reduktion der renalen Endorganschädigung führt (18) (19) (20). Eculizumab und Ravulizumab binden mit hoher Affinität an C5 und verhindern so die Entstehung des terminalen Komplementkomplexes (Abb. 3).

Multiple Fallberichte sowie Phase-II-Studien haben bereits über die Wirksamkeit der terminalen Komplementblockade mittels Eculizumab berichtet, welches neben Ravulizumab der einzige aktuell in der Schweiz zugelassene Komplementinhibitor zur Therapie des aHUS ist (8). Ravulizumab besitzt eine längere Wirkdauer und wird bei Erwachsenen nach zwei Gaben im Abstand von zwei Wochen alle acht Wochen verabreicht, was für die Patienten häufig eine deutliche Vereinfachung der Therapie im Vergleich zu den zweiwöchentlichen Gaben von Eculizumab ist. Es ermöglicht somit eine Reduktion der durchschnittlichen jährlichen Infusionszeit und Behandlungsdauer auf höchstens sechs Stunden und somit deutlich mehr Flexibilität und ein Gewinn an Lebensqualität für die betroffenen Patienten (21). Therapieansprechen und -erfolg mit Senkung der Akutkomplikationen sowie das Verhindern einer chronischen Niereninsuffizienz sind jedoch massgeblich vom Zeitpunkt der korrekten Einordnung des Krankheitsbildes abhängig. Es besteht ein begrenztes therapeutisches Fenster, bevor irreversible renale Schäden eintreten (18).
Prognose
Eine Metaanalyse aus klinischen Fallberichten der letzten zehn Jahre zu Therapieansprechen bei aHUS konnte eine statistisch signifikante Reduktion der Mortalität durch die Gabe von Eculizumab zeigen (2.3 vs. 8.8 %, p = 0.045) (22). Nachweislich kann eine vollständige Hemmung der terminalen Komplementaktivität erzielt werden sowie der Erhalt beziehungsweise die Verbesserung der betroffenen Organfunktionen. Aktuell existiert keine klinische Leitlinie über die empfohlene Dauer der medikamentösen Komplementhemmung. Rezidive der Erkrankung sind insbesondere in den ersten Monaten nach Pausierung der Therapie häufig, können aber lebenslang auftreten. Beobachtungsstudien sowie das französische Nationalregister für aHUS-Erkrankungsfälle zeigten bei durchschnittlicher Therapiesistierung nach 18 Monaten Rückfälle bei bis zu 30 % der beobachteten Patienten (19) (23). Bei näherer Differenzierung der Patientengruppe konnte jedoch gezeigt werden, dass Eculizumab bei über 50 % der untersuchten Patienten erfolgreich sistiert wurde. Dies betraf insbesondere Patienten ohne identifizierbare Mutationen (13). Ein erhöhter CD5b-9-Plasmawert wiederum galt als Hinweis für ein erhöhtes Rezidivrisiko nach Absetzen der Medikation (24).
Diskussion
Der beschriebene Patientenfall präsentiert die Fallstricke und Herausforderungen im Rahmen von Diagnostik und Management einer thrombotischem Mikroangiopathie nach Organtransplantation. Das aHUS gilt als Ausschlussdiagnose, welche das interdisziplinäre Zusammenspiel verschiedener internistischer Disziplinen erforderlich macht. Vor allem die differenzialdiagnostische Unterscheidung einer sekundären TMA und eines aHUS stellt eine besondere Herausforderung dar. Der Kostenträger besteht auf einer klaren Unterscheidung der Entitäten, um die Kosten für die teure Komplement-inhibitorische Therapie im Falle eines aHUS zu übernehmen. Dies stellt sich als besonders schwierig heraus, da Auslöser einer sekundären TMA zugleich als aggravierende Faktoren der Komplementdysregulation im Rahmen eines aHUS fungieren können. So stellt eine Organtransplantation ein hohes Risiko für die Entstehung einer sekundären TMA als auch für die überschiessende Aktivierung eines dysregulierten Komplementsystems dar. In unserem Fall sind vor allem die immunsuppressiven Medikamente wie der mTOR-Inhibitor Everolimus und der Calcineurin-Inhibitor Tacrolimus zu nennen. Insbesondere die Kombination beider immunsuppressiven Arzneimittel nach Lungentransplantation bergen erwiesenermassen ein erhöhtes Risiko für die Entstehung einer TMA (25). Als ursächlich wird hier die Induktion der Thrombozytenaggregation durch die einzelnen Wirkstoffe sowie eine generelle endotheliale Zytotoxizität diskutiert, die in der Folge eine mikrovaskuläre Schädigung bedingen (25). Diese Medikamente sind nach Lungentransplantation für den Erhalt der Allograftfunktion lebenswichtig. Umso dramatischer stellt sich die Entscheidung dar, diese Medikamente zu reduzieren, zu pausieren oder zu wechseln. Bei dieser Unterscheidung helfen kann die genetische Testung auf eine zugrunde liegende Komplementmutation, welche der Auslöser einer aHUS-Manifestation sein kann.
Es ist an dieser Stelle wichtig zu bemerken, dass ein fehlender Mutationsnachweis der oben genannten Gene ein aHUS nicht ausschliesst, da bei einem grossen Teil der aHUS-Patienten keine spezifische Mutation nachgewiesen werden kann. Auch die Messung der Komplementaktivitätsmarker im Serum können ein aHUS nicht sicher von anderen TMA-Manifestationen unterscheiden. Zudem helfen die Resultate nicht bei der Entscheidung zur Therapieinitiierung, da hier schnell gehandelt werden muss, um eine irreversible Schädigung zu verhindern, und die Tests oft Tage bis Wochen (Komplementfaktoren) oder gar Monate benötigen (Komplementgenetik).
Sridharan et al. testeten zur Diagnose des aHUS neun verschiedene Komplementmarker (CH50, AH50, C3, C4, Faktor B, Faktor H, C4d, Bb sowie sC5b-9) (26). Obwohl die Sensitivität der Kombination dieser Marker annähernd 100 % darstellt, sollten dieselben aufgrund niedriger Spezifität nur in enger Zusammenschau der weiteren klinischen Befunde interpretiert werden. Die Kenntnis einer Komplement-aktivierenden Mutation ist essenziell hinsichtlich der Entscheidungen zur Therapiedauer (13). Aktuell besteht kein gemeinsamer Konsens über die zu empfehlende Dauer der Komplementinhibition bei aHUS. Während sich die Therapie als äusserst effektiv hinsichtlich Milderung der Krankheitsaktivität erwiesen hat, sind gleichzeitig, mitunter schwerwiegende Nebenwirkungen wie das Risiko einer Meningokokkenmeningitis und die hohen Kosten von bis zu 600 000 Euro pro Jahr und Patient zu bedenken (27) (28).
Eculizumab gilt als eines der teuersten Medikamente überhaupt auf dem pharmazeutischen Markt. Die Beobachtung, dass Rezidive nach Therapiestopp häufiger bei Patienten mit besonders hohen Spiegeln des terminalen Komplementkomplexes auftreten sowie dass ein positives Ergebnis der untersuchten prädisponierenden Gene im Sinne eines Mutationsnachweises ebenfalls ein erhöhtes Risiko für ein Rezidiv darstellt, verdeutlicht die Wichtigkeit der Genanalyse und zeigt gleichzeitig die Notwendigkeit, das therapeutische Sistieren der Komplementinhibition als individuelle Einzelfallentscheidung zu behandeln (29) (30). Der lange diagnostische Weg zur final persistierenden Ausschlussdiagnose eines aHUS sowie dessen aussergewöhnlich teure Therapie sind für die beobachtete Zurückhaltung der Krankenkassen verantwortlich. Schlussendlich zeigt die Tatsache, dass die behandelnde Ärzteschaft den nationalen TMA-Expertenbeirat hinzuziehen musste, um das ablehnende Votum des Vertrauensarztes der Krankenversicherung zu überstimmen und die erfolgreiche Therapie mit Eculizumab fortzuführen, wie wichtig ein interdisziplinäres Zusammenspiel bei der Diagnostik und Therapie dieses seltenen Syndroms ist.
Abkürzungen
ADAMTS13 A-Disintegrin and metalloprotease with thrombospondin- 1-like domains
aHUS atypisches hämolytisches urämisches Syndrom
AKI acute kidney injury
ANA/ANCA Antinukleäre Antikörper/Antineutrophile cytoplasmatische Antikörper
aTTP erworbene thrombotisch-thrombozytopenische Purpura
C3, 4, 5 Komplementfaktor 3, 4, 5
CD20 Cluster of Differentiation 20 bei B-Lymphozytenantigen
CD46 Cluster of Differentiation 46 (Cofaktor für Komplementfaktor I)
CFB Komplementfaktor B
CFH-H3 CFH-spezifischer Haplotyp
CFH Komplementfaktor H
CFHR Komplementfaktor-H-related-protein
CFI Komplementfaktor-I
CNI Calcineurin-Inhibitor
CTD-ILD connective tissue disease-associated interstitial lung disease
DAT Direkter Antiglobulintest (auch direkter Coombs-Test)
DGKE Diacylglycerol Kinase Epsilon
FeNa Fraktionelle Natriumexkretion
LDH Lactatdehydrogenase
MAC Membranangriffskomplex
MBL Mannose-bindendes Lektin
MCP Membrancofaktorprotein
MMF Mycophenolat mofetil
mTOR mammalian target of Rapamycin
sMAC löslicher Membranangriffskomplex
STEC-HUS Shigatoxin bildende E.-coli-Stämme hämolytisch urämisches Syndrom
SZT Stammzelltransplantation
THBD Thrombomodulin
TMA Thrombotische Mikroangiopathie
TTP Thrombotisch-thrombozytopenische Purpura
vWF von-Willebrand-Faktor
Klinik für Pneumologie
Universitätsspital Zürich, Zürich
Medizinische Fakultät
RWTH Aachen
Oberärztin
Klinik für Nephrologie
Universitätsspital Zürich
stephanie.damm@usz.ch
Klinik für Pneumologie
Universitätsspital Zürich
Rämistrasse 100
8091 Zürich
silvia.ulrich@usz.ch
Oberärztin meV
Klinik für Pneumologie
Universitätsspital Zürich
carolin.steinack@usz.ch
Oberarzt
Klinik für Pneumologie
Universitätsspital Zürich
thomas.gaisl@usz.ch
Chefarzt Nephrologie
Kantonsspital Baden (KSB)
Klinik für Nephrologie
Universitätsspital Zürich
Klinik für Pneumologie
Leitung Rauchstoppsprechstunde
Universitätsspital Zürich
Rämistrasse 100
8091 Zürich
mace.schuurmans@usz.ch
Oberarzt
Klinik für Pneumologie
Universitätsspital Zürich
Rämistrasse 100
CH-8091 Zürich
maurice.roeder@usz.ch
Die Autorinnen und Autoren haben keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Gäckler A, Witzke O. Thrombotische Mikroangiopathie. Der Nephrologe. 2021;16(2):113-23.
2. Miller DP, Kaye JA, Shea K, Ziyadeh N, Cali C, Black C, Walker AM. Incidence of Thrombotic Thrombocytopenic Purpura/Hemolytic Uremic Syndrome. Epidemiology. 2004;15(2):208-15.
3. D.R. Terrell LAW. The incidence of thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: all patients, idiopathic patients, and patients with severe ADAMTS-13 deficiency. Journal of thrombosis and haemostasis. July 2005;3(7).
4. McFarlane PA, Bitzan M, Broome C, Baran D, Garland J, Girard LP, et al. Making the Correct Diagnosis in Thrombotic Microangiopathy: A Narrative Review. Can J Kidney Health Dis. 2021;8:20543581211008707.
5. George JN. Management of immune thrombocytopenia–something old, something new. N Engl J Med. 2010;363(20):1959-61.
6. Holle J, Lange-Sperandio, B., Mache, C., Oh, J., Pape, L., Schaefer, F., Vester, U., Weber, L. T. and Müller, D. Hämolytisch-urämisches Syndrom im Kindes- und Jugendalter. Monatsschrift Kinderheilkunde. 2017;165.
7. Yan K, Desai K, Gullapalli L, Druyts E, Balijepalli C. Epidemiology of Atypical Hemolytic Uremic Syndrome: A Systematic Literature Review. Clin Epidemiol. 2020;12:295-305.
8. Loirat C, Frémeaux-Bacchi V. Atypical hemolytic uremic syndrome. Orphanet J Rare Dis. 2011;6:60.
9. Campistol JM, Arias M, Ariceta G, Blasco M, Espinosa L, Espinosa M, et al. An update for atypical haemolytic uraemic syndrome: diagnosis and treatment. A consensus document. Nefrologia. 2015;35(5):421-47.
10. Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, Bettinaglio P, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108(4):1267-79.
11. Ratgeber R. EHEC Erkrankung – Vorkommen. Epidemiologisches Bulletin des Robert Koch Instituts. 2023;Ausgabe 36/2023.
12. Albrecht S, Kamla CE, Schönermarck U, Wassilowsky D. [Thrombotic microangiopathies in surgical intensive care medicine with special consideration of atypical hemolytic uremic syndrome]. Anaesthesiologie. 2023;72(1):3-12.
13. Knoop M, Haller H, Menne J. [Human genetics in atypical hemolytic uremic syndrome-its role in diagnosis and treatment]. Internist (Berl). 2018;59(8):799-804.
14. Nester CM, Barbour T, de Cordoba SR, Dragon-Durey MA, Fremeaux-Bacchi V, Goodship TH, et al. Atypical aHUS: State of the art. Mol Immunol. 2015;67(1):31-42.
15. Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5(10):1844-59.
16. Fremeaux-Bacchi V, Fakhouri F, Garnier A, Bienaimé F, Dragon-Durey MA, Ngo S, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol. 2013;8(4):554-62.
17. Thompson GL, Kavanagh D. Diagnosis and treatment of thrombotic microangiopathy. Int J Lab Hematol. 2022;44 Suppl 1(Suppl 1):101-13.
18. Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368(23):2169-81.
19. Rondeau E, Scully M, Ariceta G, Barbour T, Cataland S, Heyne N, et al. The long-acting C5 inhibitor, Ravulizumab, is effective and safe in adult patients with atypical hemolytic uremic syndrome naïve to complement inhibitor treatment. Kidney Int. 2020;97(6):1287-96.
20. Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol. 2007;25(11):1256-64.
21. Facharztmagazine R. Zulassung für Komplementinhibitor erweitert. Im Fokus Onkologie. 2021;24(1):58-.
22. Krishnappa V, Gupta M, Elrifai M, Moftakhar B, Ensley MJ, Vachharajani TJ, et al. Atypical Hemolytic Uremic Syndrome: A Meta-Analysis of Case Reports Confirms the Prevalence of Genetic Mutations and the Shift of Treatment Regimens. Therapeutic Apheresis and Dialysis. 2018;22(2):178-88.
23. Olson SR, Lu E, Sulpizio E, Shatzel JJ, Rueda JF, DeLoughery TG. When to Stop Eculizumab in Complement-Mediated Thrombotic Microangiopathies. Am J Nephrol. 2018;48(2):96-107.
24. Fakhouri F, Fila M, Hummel A, Ribes D, Sellier-Leclerc AL, Ville S, et al. Eculizumab discontinuation in children and adults with atypical hemolytic-uremic syndrome: a prospective multicenter study. Blood. 2021;137(18):2438-49.
25. Hachem RR, Yusen RD, Chakinala MM, Aloush AA, Patterson GA, Trulock EP. Thrombotic microangiopathy after lung transplantation. Transplantation. 2006;81(1):57-63.
26. Sridharan M, Go RS, Abraham RS, Fervenza FC, Sethi S, Bryant SC, et al. Diagnostic Utility of Complement Serology for Atypical Hemolytic Uremic Syndrome. Mayo Clin Proc. 2018;93(10):1351-62.
27. Mallett A, Hughes P, Szer J, Tuckfield A, Van Eps C, Cambell SB, et al. Atypical haemolytic uraemic syndrome treated with the complement inhibitor eculizumab: the experience of the Australian compassionate access cohort. Internal Medicine Journal. 2015;45(10):1054-65.
28. (ON): APCCINosdO. Pharmacoeconomic Report: Eculizumab (Soliris): Canadian Agency for Drugs and Technologies in Health; . 2020 Oct. ;Appendix 1, Cost Comparison Table.
29. Ardissino G, Testa S, Possenti I, Tel F, Paglialonga F, Salardi S, et al. Discontinuation of eculizumab maintenance treatment for atypical hemolytic uremic syndrome: a report of 10 cases. Am J Kidney Dis. 2014;64(4):633-7.
30. Merrill SA, Brittingham ZD, Yuan X, Moliterno AR, Sperati CJ, Brodsky RA. Eculizumab cessation in atypical hemolytic uremic syndrome. Blood. 2017;130(3):368-72.
PRAXIS
- Vol. 113
- Ausgabe 9
- Oktober 2024