Fallbeschreibung
Vorgestellt werden zwei Patienten mit unterschiedlichem Verlauf bei rasch fortschreitenden Wesensveränderungen und Demenz.
Fall 1
Ein 69-jähriger Mann litt seit März 2019 an unklaren rezidivierenden Dysästhesien und einer motorischen Beinschwäche links. Eine Magnetresonanztomographie (MRI) mit Angiographie des Neurokraniums zeigte keine Hinweise auf eine Durchblutungsstörung, Ischämie oder Raumforderung. Im Sommer 2019 erfolgte bei einer notfallmässigen Vorstellung aufgrund derselben Symptomatik eine Computertomographie (CT) mit Angiographie. Hier fanden sich weiterhin keine Hinweise auf eine Ischämie, jedoch kam eine mässige, am ehesten mikroangiopathische Leukenzephalopathie zur Darstellung. In der nachfolgenden Elektroenzephalographie (EEG) zeigte sich ein bitemporaler Herdbefund rechtsbetont mit Epilepsie-verdächtigen Einzelpotenzialen. Bei Verdacht auf rezidivierende fokal-epileptische Ereignisse wurde eine anfallssupprimierende Therapie mit Levetiracetam eingeleitet. Zunächst kam es zu keinen erneuten epileptischen Ereignissen. Neu wurde seitens der Ehefrau eine depressive Symptomatik beschrieben. Ein Montreal-Cognitive-Assessment(MoCA-) -Test im Januar 2020 fiel mit 18/30 Punkten pathologisch aus, sodass eine demenzielle Entwicklung vermutet wurde. Die empfohlene neuropsychologische Testung wurde nicht durchgeführt. Im Sommer 2020 wurde der Patient schliesslich bei raschem Fortschreiten der demenziellen Entwicklung und Allgemeinzustandsverschlechterung bei uns auf der neurologischen Station hospitalisiert. In der Lumbalpunktion fanden sich nun neben einer Schrankenstörung eine stark erhöhte Proteinzahl von 3600 mg/l (Normwert: 150–450 mg/l) und eine Erhöhung der Zellzahl von 21 mononukleären Zellen/μl (Normwert: < 4 Zellen/μl). Das Tau-Protein war mit 1266 pg/ml (Normwert: < 445 pg/ml) ebenfalls erhöht und das Beta-Amyloid-Protein mit 282 pg/ml (Normwert: > 375 pg/ml) erniedrigt. Das Phospho-Tau-Protein lag mit 35.5 pg/ml im Normalbereich (Normwert: < 61 g/ml). Diese Konstellation im Liquor mit erhöhtem Tau-Protein und erniedrigtem Beta-Amyloid-Protein kann sowohl für eine Demenzform wie Alzheimer aber auch eine inflammatorische cerebrale Amyloidangiopathie (Englisch: cerebral amyloid angiopathy related inflammation; CAA-RI) sprechen. Ein MRI des Neurokraniums mit Kontrastmittel zeigte eine ausgeprägte superfizielle Siderose, eine frische subarachnoidale Sickerblutung und ein Ödem frontal beidseits (Abb. 1). Diese Befunde waren passend für eine CAA-RI. Eine zentrale Manifestation einer systemischen Vaskulitis erschien bei negativen antinukleären Antikörpern (ANA) und antineutrophilen zytoplasmatischen Antikörpern (ANCA) unwahrscheinlich. Eine serologische Testung des sauren Gliafaserproteins (Englisch: glial fibrillary acidic protein; GFAP) und der Neurofilament-Leichtketten (NFL) erfolgte nicht. Insgesamt wurde nun die Verdachtsdiagnose einer inflammatorischen cerebralen Amyloidangiopathie gestellt. Eine Steroidstosstherapie mit Methylprednisolon intravenös 1 g/Tag über 5 Tage mit anschliessender Erhaltungsdosis mit Prednisolon von 1 mg/kg Körpergewicht wurde eingeleitet. Hierunter kam es zu keiner signifikanten klinischen Besserung. Im MRI des Neurokraniums mit Kontrastmittel nach 1 Woche konnte noch keine wesentliche Befundänderung dokumentiert werden. Die zuvor in der Liquorpunktion erhöhten Werte waren rückläufig (Proteinzahl 465 mg/l, 12 mononukleäre Zellen/μl).
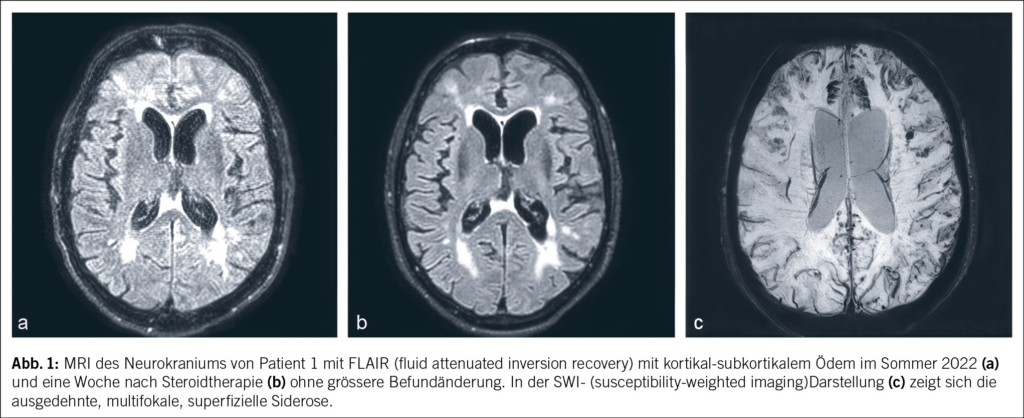
Es erfolgte ein Übertritt in die neurologische Rehabilitation über 1 Monat. Dort wurde vor allem die antiepileptische und psychiatrische Therapie weiter angepasst. Insgesamt blieb der Patient im Antrieb stark gemindert, selektiv mutistisch und konnte keine Handlungen ohne Handlungsplan durchführen. Der Patient wurde anschliessend bei Verdacht auf ein hypoaktives Delir direkt in die Psychiatrie zur weiteren medikamentösen Einstellung überwiesen. Aufgrund einer akuten Vigilanzminderung, Fieber, Tachykardie und Meningismus wurde er nach 10 Tagen wieder ins Spital überwiesen. Laborbefunde zeigten nun ein isoliert erhöhtes CRP. Im CT-Schädel fand sich eine diskrete akute Subarachnoidalblutung im rechten Temporallappen. Neben einer wieder erhöhten Proteinzahl von 1398 mg/l (Normwert: 150–450 mg/l) war die Liquoruntersuchung inklusive einer Multiplex-PCR (polymerase chain reaction) für Meningitis-Erreger unauffällig. Blutkulturen blieben ohne Wachstum. Entsprechend wurde die auf dem Notfall etablierte antimikrobielle Therapie mit Ceftriaxon und Aciclovir wieder ausgesetzt. Auch ein Therapieversuch mit Levetiracetam, welcher bei Verdacht auf einen nicht konvulsiven Status epilepticus gestartet wurde, zeigte keine Symptombesserung, und die Therapie wurde im Verlauf abgebrochen. Da eine Hochdosis-Steroidtherapie bei der ersten Hospitalisation ohne Besserung auf die rasch fortschreitende demenzielle Entwicklung blieb, wurde auf einen zweiten Zyklus verzichtet.
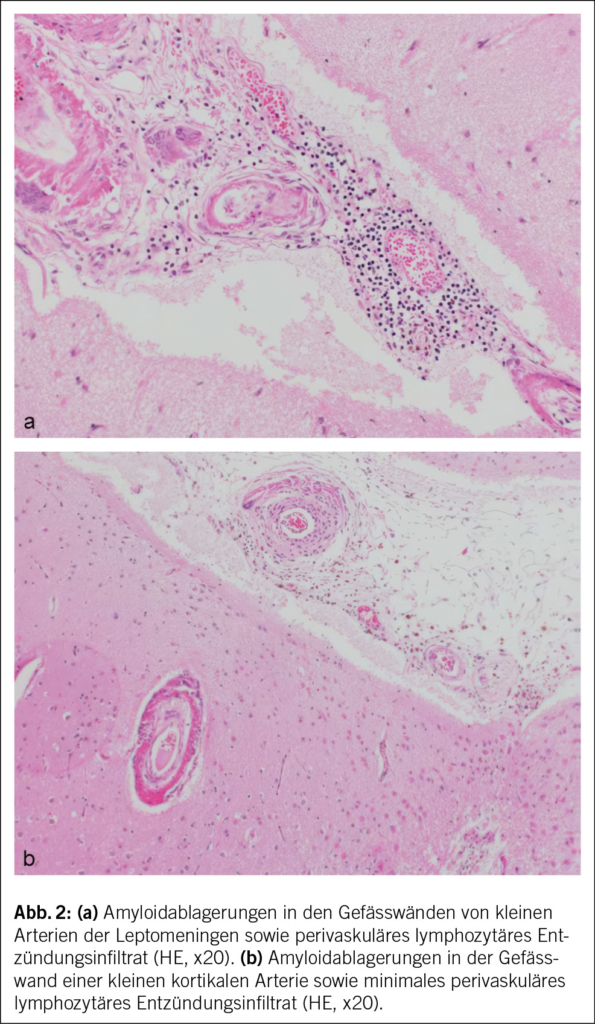
In Rücksprache mit den Angehörigen wurde bei infauster Prognose schliesslich eine palliative Therapie eingeleitet, und der Patient verstarb auf der Palliativstation am 10. Hospitalisationstag Ende Oktober 2020. Die Autopsie bestätigte schliesslich die Diagnose einer CAA-RI aufgrund einer deutlichen Beteiligung der Gefässe in den Leptomeningen und im Cortex, mit fokal geringer begleitender T-lymphozytärer Entzündungsreaktion sowie zahlreichen kortikalen Infarkten und perivaskulären Mikroblutungen (Abb. 2). Die CAA-RI führte über Gefässverschlüsse zu einer diffusen vaskulär-ischämischen Leukenzephalopathie und mehreren frischen und älteren, nicht raumfordernden Subarachnoidalblutungen. Das gesamte Bild sprach für eine CAA-RI und nicht für eine Beta-Amyloid-assoziierte Angiitis (ABRA), welche ein ähnliches histologisches Bild zeigt, jedoch mit ausgeprägter vaskulitischer Komponente und fibrinoiden Gefässwandnekrosen. Es zeigten sich dazu deutliche Alzheimer-assoziierte Veränderungen mit Tau-positiven Neurofibrillendegeneraten und neuritischen Plaques im Hippocampus und Temporal-Cortex und vereinzelt Tangles im frontalen Cortex (Stadium IV nach Braak und Braak, CERAD-3 und ABC-Score A3 B2 C3), dies bei deutlicher äusserer und innerer Hirnatrophie. Zudem fand sich überraschenderweise ein Hämatom der vorderen Bauchwand mit einem Volumen von ca. 1500 ml, und es wurde eine Lobärpneumonie beider Lungen diagnostiziert. Ob die Pneumonie ursächlich für die bislang unklare CRP-Erhöhung war oder die Infektion in den letzten Lebenstagen auf der Palliativstation entwickelt wurde, konnte nicht abschliessend geklärt werden. Die Ätiologie des Hämatoms blieb letztlich unklar. Histologisch fanden sich hier keine Hinweise auf eine Gefässmalformation, Amyloidangiopathie oder Vaskulitis. Ein stumpfes Trauma wurde in der aktuellen Hospitalisation nicht beobachtet, der Patient war nicht antikoaguliert, und während der letzten Hospitalisation erfolgten keine Massnahmen abdominal (z. B. Insulin- oder Heparinspritzen). Nicht eruierbar waren mögliche Vorfälle in den vorangehenden Institutionen. Abdominale Blutungen bei Patienten mit CAA-RI wurden bisher nicht als Assoziation beschrieben. Als Todesursache wurde ein kardiopulmonales Versagen aufgrund der Pneumonie sowie Volumenmangel aufgrund des Hämatoms angegeben.
Fall 2
Eine 74-jährige Patientin wurde im Januar 2022 mit seit einigen Wochen fortschreitender Verwirrtheit, Verlangsamung sowie kognitiver Beeinträchtigung aus einem peripheren Spital in unsere Notaufnahme überwiesen. Ausserdem bestand ein progredienter Mutismus, welcher im Rahmen einer Trauersituation nach dem Tod des Ehemannes 1 Monat zuvor aufgrund von Covid-19 angesehen wurde. Die Patientin selbst litt an Covid-19 mit milden Symptomen. Nun zeigte das MRI des Neurokraniums eine ausgedehnte Leukenzephalopathie, ein vasogenes Ödem und mehrere Mikroblutungen (Abb. 3). Die Ergebnisse des Liquors waren negativ für Treponema pallidum und Lyme-Borreliose, ebenso eine Multiplex-PCR für Meningitis-Erreger. Die Immunphänotypisierung im Liquor, welche bei Verdacht auf ein Lymphom durchgeführt wurde, war negativ für B-Zell- und T-Zell-Neoplasien. Eine Bestimmung im Liquor von Beta-Amyloid, Tau-Protein und Phospho-Tau-Protein als Demenzmarker sowie Neurofilament-Leichtketten (NFL) als Marker bei Multipler Sklerose erfolgte nicht. Ebenso wurden die Biomarker GFAP und NFL serologisch nicht untersucht. Es wurde ein EEG angefertigt. Neben moderaten Allgemeinveränderungen und bifrontaler fokaler Verlangsamung fanden sich Epilepsieverdächtige Einzelpotenziale rechtshemisphärisch, welche unter Levetiracetam-Therapie nach 1 Woche abnahmen. Klinisch kam es jedoch noch zu keiner objektivierbaren Verbesserung. Die Patientin erzielte im MoCa-Test 12/30 Punkte, was für eine starke kognitive Beeinträchtigung steht. Schliesslich wurde eine Biopsie des Hirngewebes entnommen. Diese zeigte Veränderungen im Zusammenhang mit der Alzheimer-Krankheit mit Tau-positiven Neuronen, neurofibrillären Tangles und eine grosse Menge an Beta-Amyloid-Plaques sowie Amyloidablagerungen an den Gefässwänden (Abb. 4). Zusätzlich fanden sich subarachnoidal Zeichen einer wenige Tage alten Einblutung. Aufgrund der perivaskulären Entzündung wurde schliesslich eine cerebrale Amyloidangiopathie-assoziierte Entzündung (CAA-RI) als am wahrscheinlichsten angesehen. Eine Therapie mit Methylprednisolon intravenös 1 g/Tag gefolgt von oralem Prednisolon mit 1 mg/kg Körpergewicht wurde eingeleitet. Nach der Entlassung erfolgte eine stationäre neurologische Rehabilitation. Hier kam es zu einer klinischen Verbesserung mit jedoch relevanten Einschränkungen in Bezug auf die täglichen Routinen und das Kurzzeitgedächtnis. Die Prednisolon-Dosis wurde langsam reduziert.
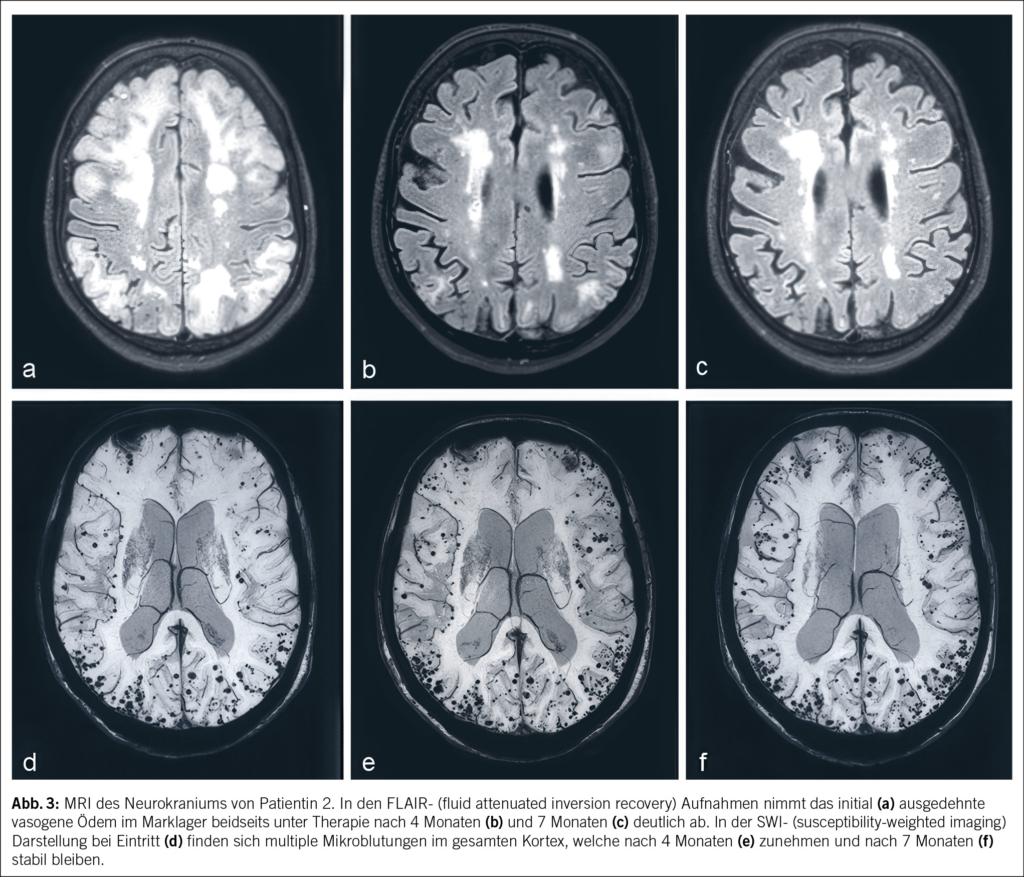
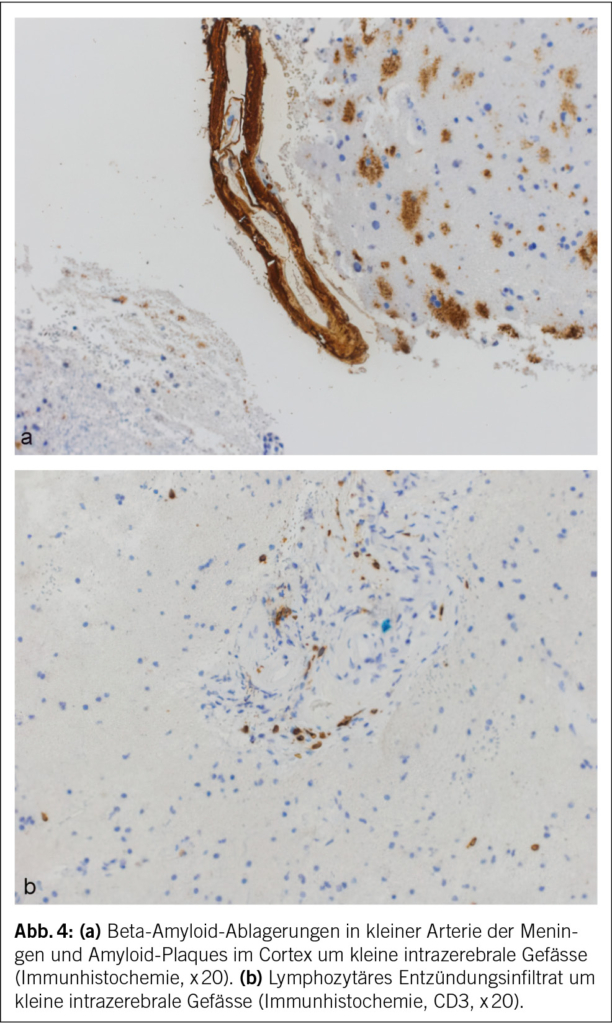
In den Verlaufskontrollen bis August 2022 nahmen die Ödemareale im MRI des Gehirns weiter ab, und es fanden sich keine neuen Läsionen. Die multiplen Mikroblutungen supratentoriell beidseits und vereinzelt infratentoriell blieben stationär. Klinisch zeigte die Patientin weiter eine Besserung mit weniger anhaltenden Einschränkungen. Im MoCA-Test wurden 3 Monate nach Diagnosestellung und Therapiebeginn 15/30 Punkte und 7 Monate nach Diagnose 25/30 Punkte erzielt. Im November 2023 wurde eine neuropsychologische Testung durchgeführt, da die Patientin die Fahrtauglichkeit wieder anstrebte. Hier konnte eine Verbesserung der Grundaktivierung, allerdings aber auch eine relevante Verschlechterung der Aufmerksamkeitsteilung, objektiviert werden. Die kognitiven Defizite umfassten fronto-temporo-parietale Ausfälle, und es bestand ein unveränderter Unterstützungsbedarf im Alltag. Die Fahreignung konnte aus neuropsychologischer Sicht weiterhin nicht bestätigt werden. Auch wurde aufgrund der Befunde des letzten MRI des Gehirns von 2022 von keiner weiteren Regredienz der kognitiven Defizite ausgegangen.
Diagnose und Kommentar
Die inflammatorische cerebrale Amyloidangiopathie (CAA-RI) ist eine seltene Erkrankung und potenziell reversibel. Sie gehört zu den cerebralen Amyloidangiopathien (CAA), bei denen es zu einer Ablagerung von Beta-Amyloid-Peptiden in den vorwiegend kleineren cerebralen Arterien kommt (1). Hierdurch entstehen degenerative Veränderungen, Gefässverschlüsse, Mikroaneurysmen, welche schliesslich zu einer diffusen vaskulär-ischämischen Leukenzephalopathie und Hirnblutungen führen. Die CAA ist für ca. 20 % aller intrazerebralen Blutungen verantwortlich (1).
Die Amyloidablagerungen sind bei einem Teil der Patienten mit einer Entzündung der Gefässwand vergesellschaftet, was schliesslich zu einem multifokalen Marklagerödem führt. Insgesamt zeigen diese Veränderungen im MRI ein typisches Bild, welche für die Diagnosestellung einer CAA-RI wesentlich sind. Die Veränderungen lassen sich vor allem in der FLAIR-Sequenz (fluid attenuated inversion recovery) und bei der SWI (Suszeptibilitätsgewichtete Bildgebung) feststellen (2, 3). Dazu gehören Mikroblutungen, eine kortikale superfizielle Siderose und eine asymmetrische fleckförmige oder konfluierende Leukenzephalopathie, welche den angrenzenden Kortex und das subkortikale Marklager miteinbeziehen können. Ebenfalls kann sich als Zeichen der entzündlichen Reaktion ein vasogenes Ödem in der ADC- (apparent diffusion coefficient) Wichtung präsentieren (2, 4, 5). Es wurden die sogenannten modifizierten Boston-Kriterien entwickelt, welche auf eine gute Sensitivität und Spezifität geprüft wurden (6) (Tab. 1) und bei der Diagnosesicherung helfen. Zusätzliche klinische Diagnosekriterien sind ein akuter/subakuter Symptombeginn, Alter über 55 Jahre, Symptome wie Kopfschmerzen, Wesensveränderungen, kognitive Defizite oder fokal neurologische Defizite oder epileptische Anfälle. Andere Ursachen (z. B. infektiös oder paraneoplastisch) müssen ausgeschlossen werden. Sind alle diese Kriterien erfüllt, gilt eine CAA-RI als wahrscheinlich. Zur definitiven Diagnosesicherung wird eine histologische Bestätigung im Rahmen einer Autopsie benötigt, wobei sich neben frischen und alten Ischämien und Einblutungen auch entzündliche, perivaskuläre Veränderungen ohne Gefässbeteiligung finden lassen. Hier kann eine Unterscheidung zur Beta-Amyloid-assoziierten Angiitis (ABRA) gemacht werden, welche ausgeprägtere vaskulitische Veränderungen und fibrinoide Gefässwandnekrosen zeigt. Diese erheblichen Zerstörungen des Hirnparenchyms direkt durch invasive zytotoxische T-Lymphozyten und indirekt durch vaskulitische oder begleitthrombotische Gefässverschlüsse bedingen eine stärkere Immunsuppression als bei der CAA-RI. Teils wird in der Literatur jedoch die ABRA synonym zur CAA-RI genannt. Ob eine Histologie zur Diagnosestellung einer CAA-RI immer zwingend ist, steht aktuell immer noch zur Diskussion. Eine genaue Diagnose hat jedoch teils therapeutische Konsequenzen. Vor allem bei fehlendem Therapieansprechen sollte eine Biopsie angestrebt werden.
![]()
Männer und Frauen sind etwa gleich häufig von einer CAA-RI betroffen (7). Das durchschnittliche Erkrankungsalter liegt bei ca. 67 Jahren. Tritt die Erkrankung bei jüngeren Personen auf, kann eine seltene familiäre Form einer CAA-RI in Betracht gezogen werden (8). Es werden monophasische, schubförmige und primär progrediente Verlaufsformen beschrieben. Über 70 % der Patienten mit CAA-RI sind homozygot für das Apolipoprotein-E-ε4-Allel (ApoE-ε4), jedoch nur < 5 % der Patienten mit CAA (7, 9, 10). ApoE-ε4 ist ebenfalls assoziiert mit der Alzheimer-Erkrankung. Eine Überlappung von Alzheimer mit CAA-RI wird auch in Autopsiestudien beschrieben. Typischerweise sind bei der CAA-RI die Proteine und Zellzahl im Liquor erhöht, das Beta-Amyloid ist erniedrigt.
Differenzialdiagnostisch zur CAA-RI sind neben der ABRA und der primären ZNS-Angiitis (primary angiitis of the central nervous system; PACNS) unter anderem das posteriore reversible Enzephalopathie-Syndrom (PRES), eine progrediente multifokale Leukenzephalopathie (PML), Neurosarkoidose, Creutzfeldt-Jakob-Krankheit oder eine Herpes-simplex-Enzephalitis zu nennen (11–13). Zunehmend häufiger werden Autoimmunenzephalitiden diagnostiziert, welche ebenfalls einen subakuten kognitiven Abbau, eine langsame Bewusstseinstrübung und epileptische Anfälle zeigen können. Weitere gut behandelbare und daher relevante Differenzialdiagnosen sind die Steroid-responsive Enzephalopathie assoziiert mit autoimmuner Thyroiditis (SREAT, Hashimoto-Enzephalopathie), eine Riesenzellarteriitis und die superfizielle Siderose des Zentralnervensystems (14–16).
Die CAA-RI zeigt ein gutes Ansprechen auf Kortikosteroide, worunter es zu einer klinischen Besserung und Regredienz der radiologischen Befunde kommt. Am geringsten kommt es zu einer Verbesserung der kognitiven Symptome. Die rechtzeitige und frühe Therapieeinleitung ist essenziell, um ein möglichst gutes Outcome zu erreichen. Bei Nichtansprechen auf die Steroidtherapie oder bei einer Progression oder Rezidiv (ca. 25 % der Fälle [7]) wurden teils erfolgreich auch andere immunsuppressive Therapien (z. B. Methotrexat, Cyclophosphamid, Immunglobuline) eingesetzt (13, 17). Die Therapiedauer mit einem langsamen Ausschleichen der Kortikosteroide ist eine Einzelfallentscheidung und vom Verlauf abhängig.
Die Diagnose einer CAA-RI gestaltet sich aufgrund des unterschiedlichen, wenig spezifischen klinischen Bildes oft als sehr schwierig. Bei entsprechenden Symptomen soll aber auch an eine CAA-RI gedacht werden und entsprechend die weitere Diagnostik verfolgt werden. Wichtige Säulen bilden das MRI und, sofern möglich, die Histologie.
Medizinisches Zentrum gleis d
Gürtelstrasse 46
7000 Chur
d.imhoff@mez-chur.ch
Die Autorenschaft hat keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
1. Vinters HV. Cerebral amyloid angiopathy. A critical review. Stroke 1987;18:311-324
2. Kinnecom C, Lev MH, Wendell L, et al. Course of cerebral amyloid angiopathy-related inflammation. Neurology 2007;68:1411-1416
3. Cheng AL, Batool S, McCreary CR, et al. Susceptibility-weighted imaging is more reliable than T2*-weighted gradient-recalled echo MRI for detecting microbleeds. Stroke 2013;44:2782-2786
4. Martucci M, Sarria S, Toledo M, et al. Cerebral amyloid angiopathy-related inflammation: imaging findings and clinical outcome. Neuroradiology 2014;56:283-289
5. Raghavan P, Looby S, Bourne TD, et al. Cerebral amyloid angiopathy-related inflammation: A potentially reversible cause of dementia with characteristic imaging findings. J Neuroradiol. 2016;43:11-17
6. Auriel E, Charidimou A, Gurol ME, et al. Validation of Clinicoradiological Criteria for the Diagnosis of Cerebral Amyloid Angiopathy-Related Inflammation. JAMA Neurol. 2016;73:197-202
7. Chung KK, Anderson NE, Hutchinson D, et al. Cerebral amyloid angiopathy related inflammation: three case reports and a review. J Neurol Neurosurg Psychiatry. 2011;82:20-26
8. Zhang-Nunes SX, Maat-Schieman ML, van Duinen SG, et al. The cerebral beta-amyloid angiopathies: hereditary and sporadic. Brain Pathol. 2006;16:30-39
9. Schaumberg J, Trauscheid M, Eckert B, et al. Zerebrale Amyloidangiopathie assoziiert mit Inflammation (Cerebral amyloid angiopathy associated with inflammation). Nervenarzt. 2018;89:682-691
10. Charidimou A, Gang Q, Werring DJ. Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. J Neurol Neurosurg Psychiatry. 2012;83:124-137
11. Moussaddy A, Levy A, Strbian D, et al. Inflammatory Cerebral Amyloid Angiopathy, Amyloid-ss-Related Angiitis, and Primary Angiitis of the Central Nervous System: Similarities and Differences. Stroke 2015;46:e210-213
12. Danve A, Grafe M, Deodhar A. Amyloid beta-related angiitis–a case report and comprehensive review of literature of 94 cases. Semin Arthritis Rheum. 2014;44:86-92
13. Wu JJ, Yao M, Ni J. Cerebral amyloid angiopathy-related inflammation: current status and future implications. Chin Med J (Engl). 2021;134:646-654
14. Fellinger M, Friedrich F, Vafai Tabrizi F, et al. Hashimoto-Enzephalopathie. Psychopraxis. Neuropraxis. 2017; 20, 66-71
15. Zeidler M, Hughes T, Zeman A. Confused by arteritis. The Lancet 2000;355:374-375
16. Chen J, Cabahug P, Edmiston T. Superficial Siderosis of the Central Nervous System: A Report of Two Cases With Spinal Pathology and a Review of the Literature. Cureus. 2024;16(5):e60486
17. Chwalisz BK. Cerebral amyloid angiopathy and related inflammatory disorders. J Neurol Sci. 2021;424:117425