Einleitung
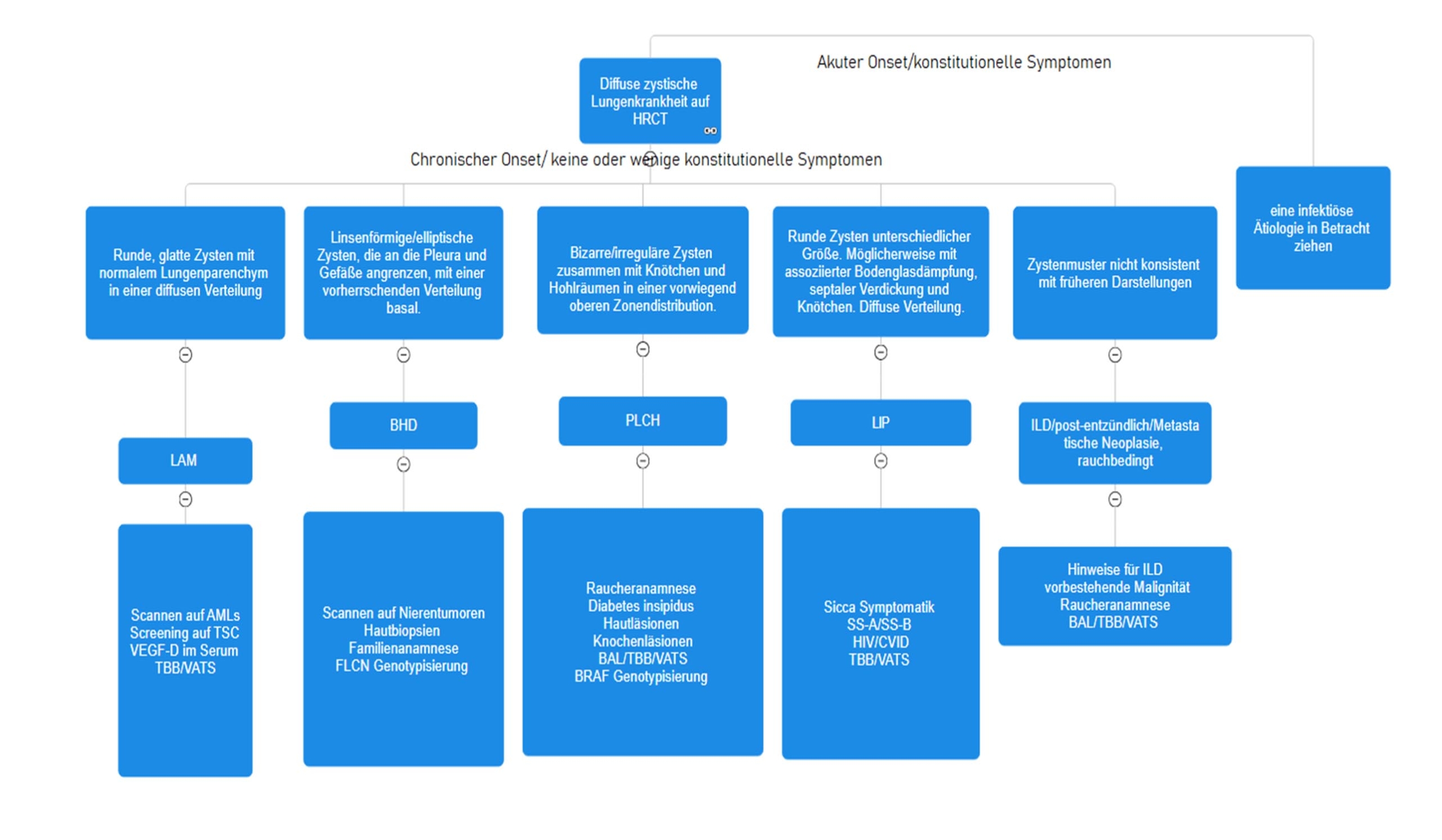
Lungenzysten zählen zu den vermehrt diagnostizierten radiologischen Zufallsbefunden, begünstigt durch die weit verbreitete Verfügbarkeit von hochauflösenden Computertomographien (HRCT). Sie können als Teil des natürlichen Alterungsprozesses des Lungenparenchyms auftreten oder sich im Rahmen verschiedener Lungenerkrankungen manifestieren, einschliesslich postinfektiöser, maligner oder entzündlicher Genese. Bei der Konfrontation mit Lungenzysten in der HRCT ist ein systematischer Ansatz unerlässlich (Abbildung 1). Der erste Schritt besteht darin, sie von anderen umschriebenen, gering absorbierenden Bereichen zu unterscheiden, die häufig auftreten, beispielsweise Emphysem, Bronchiektasien oder Honeycombing. Anschliessend klassifiziert die Anzahl der Zysten, das Muster der Verteilung sowie das Vorhandensein von begleitenden Lungenanomalien die Zysten weiter in lokalisierte, zufällig auftretende oder diffuse zystische Lungenerkrankungen. Das Vorhandensein einer diffusen zystischen Lungenerkrankung rechtfertigt weitere Tests für eine präzise Diagnose, da die Behandlungsoptionen krankheitsspezifisch sind.
Birt-Hogg-Dubé-Syndrom
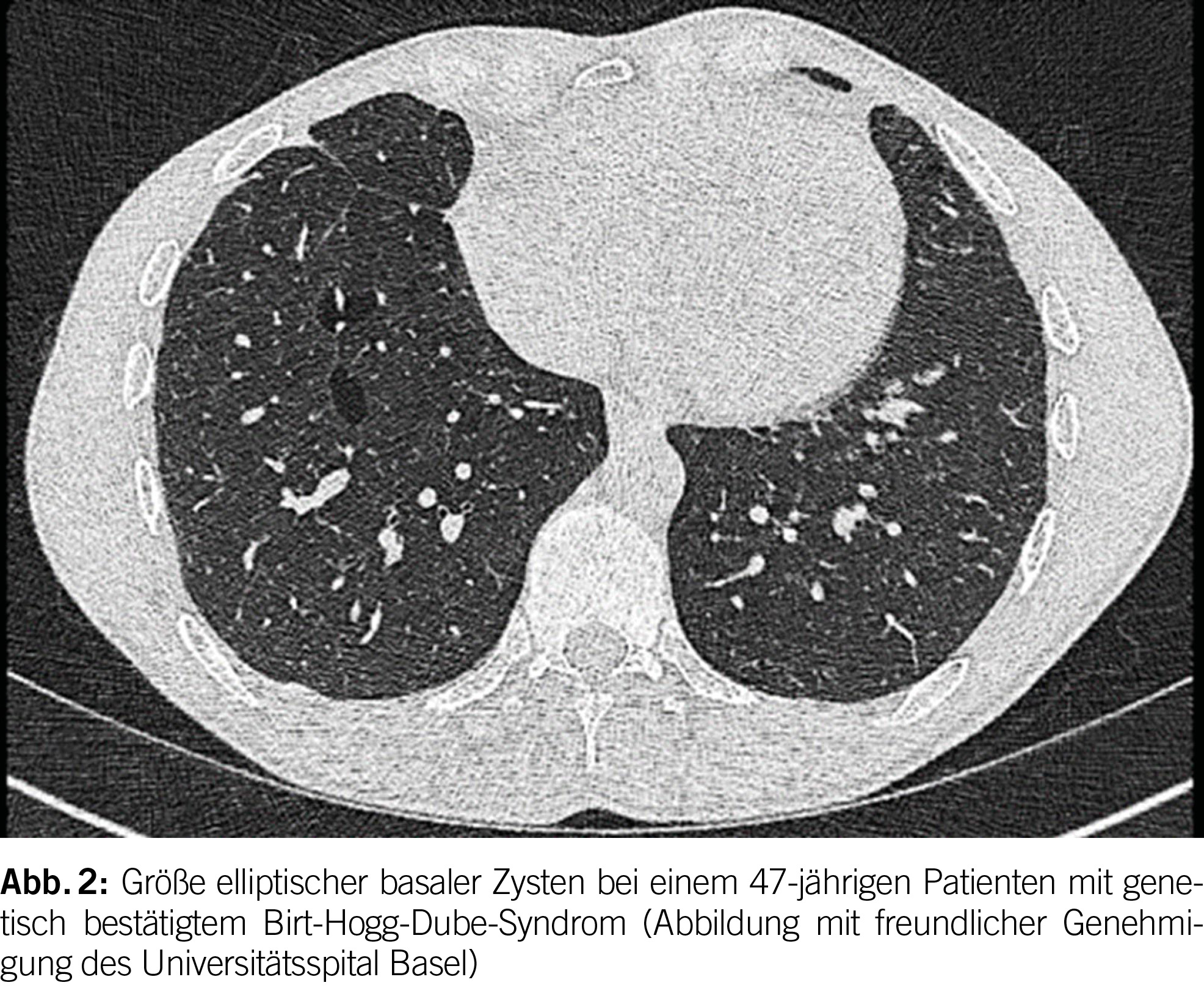
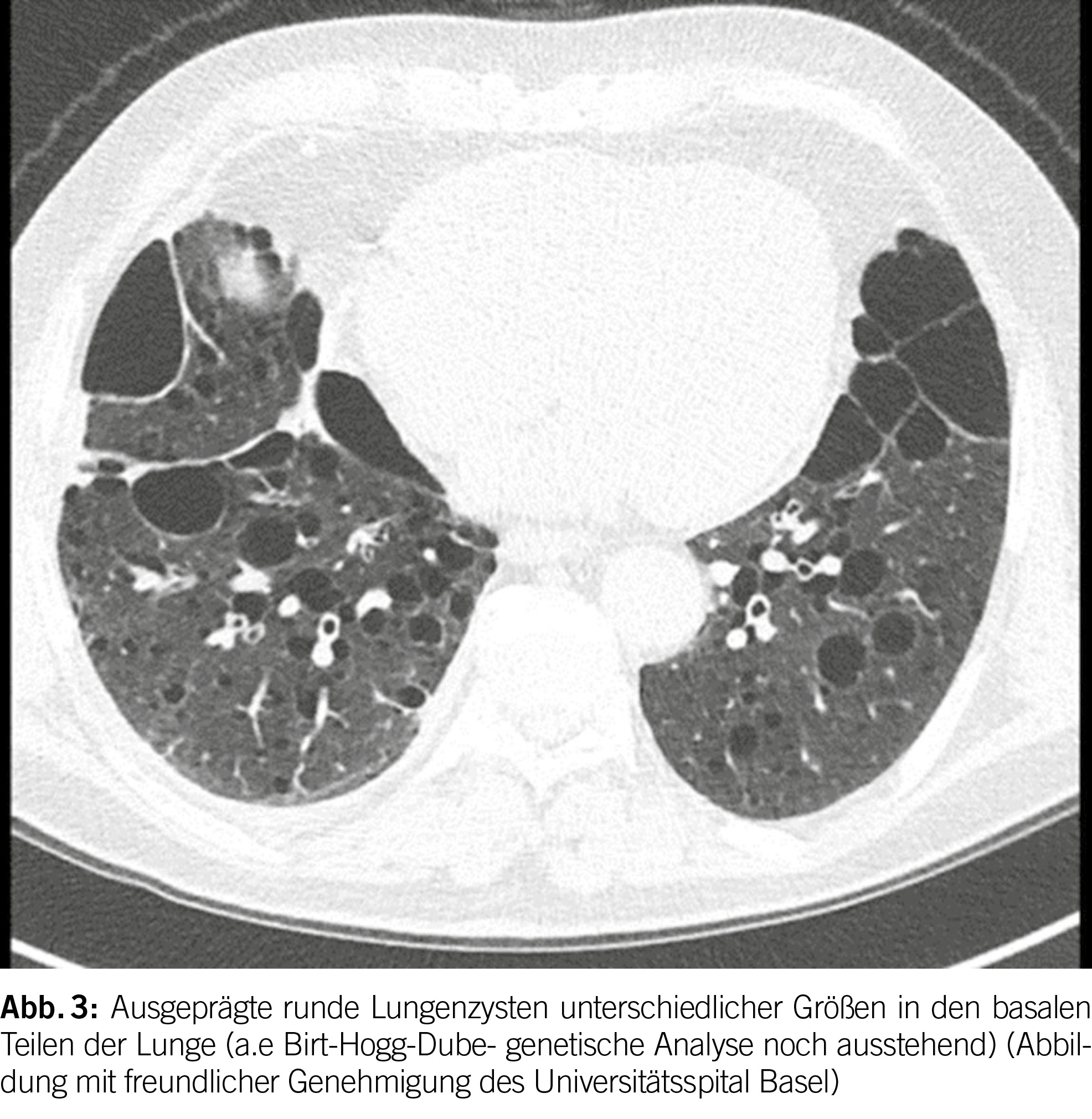
Das Birt-Hogg-Dubé Syndrom (BHD) ist eine seltene autosomal dominante Störung mit einer hohen Penetranz (geschätzt bei 90-95%) (1). Sie entsteht aufgrund von Keimbahnmutationen im Tumorsuppressorgen FLCN, das auf Chromosom 17p11.2 liegt und das Folliculin-Protein codiert. Die phänotypischen Merkmale der Krankheit umfassen eine Triade aus diffusen pulmonalen Zysten, Hautläsionen und renalen Neoplasien unterschiedlicher Histologie. Es betrifft in der Regel junge Erwachsene ohne Geschlechtspräferenz und tritt überwiegend bei Personen im Alter von 20 bis 40 Jahren auf; es kann jedoch in allen Altersgruppen auftreten. Bei BHD werden bei der Mehrheit der Patienten (ungefähr 80%) pulmonale Zysten beobachtet. Diese Zysten weisen im Vergleich zu anderen diffusen zystischen Lungenerkrankungen (DZLE) eine linsenförmige Form, größere Abmessungen und eine basale Dominanz auf (Abbildungen 2 und 3). Eine Studie zu radiologischen Merkmalen, die mit den vier Hauptursachen diffuser Lungenzysten assoziiert sind, zeigte eine deutlich höhere Inzidenz von paramediastinalen Zysten bei Personen mit BHD (2).
Die klinischen Manifestationen der Lungenbeteiligung bei BHD sind unspezifisch und werden hauptsächlich durch ein erhöhtes Risiko für die Entwicklung eines Pneumothorax charakterisiert (etwa 50-mal höher als in der Allgemeinbevölkerung) (3). So entwickeln etwa 24% der Patienten mit Lungenzysten bei BHD einen Pneumothorax mit einer sehr hohen Rezidivrate von 75% (1).
Eine Hautbeteiligung tritt bei mehr als 85% der Patienten auf und äussert sich durch die Bildung gutartiger Hauttumore wie Fibrofollikulomen, Trichodiskomen und Acrochordonen (1). Die charakteristischsten Hautläsionen bei BHD sind Fibrofollikulome, schmerzlose kleine papuläre Wucherungen, die sich allmählich über die Kopfhaut, das Gesicht, den Hals und die Brust verteilen.
Eng mit einer erhöhten Mortalität verbunden ist eine renale Beteiligung, die bei etwa 30% der Patienten mit BHD diagnostiziert wird (4). Die häufigsten Histologien umfassen Onkozytome und chromophobe Adenome, obwohl klarzellige und papilläre Karzinome ebenfalls auftreten können. Das Vorhandensein von bilateralem oder multifokalem Nierenkrebs mit frühem Beginn sollte den Verdacht auf BHD wecken.
Die Identifikation eines Pneumothorax bei jungen Personen mit einer positiven Familienanamnese bezüglich Pneumothoraces oder Nierentumoren sollte eine Untersuchung auf das Birt-Hogg-Dubé-Syndrom nach sich ziehen. Diese Evaluation umfasst eine hochauflösende Computertomographie (HRCT), eine dermatologisches Konsil mit Hautbiopsie sowie genetische Testung. Eine Lungenbiopsie ist bei typischer Konstellation respektive nachgewiesener Mutation nicht indiziert.
Die Behandlung besteht hauptsächlich in der Bewältigung von Komplikationen. Darüber hinaus ist das Screening auf renale Neoplasien entscheidend und wird in der Regel ab dem 20. Lebensjahr mit regelmässigen Screenings im Intervall von drei Jahren unter Verwendung der Magnetresonanztomographie (MRT) empfohlen (5,6).
Lymphozytäre interstitielle Pneumonie
Die lymphozytäre interstitielle Pneumonie (LIP) beschreibt eine diffuse Infiltration des Lungenparenchyms durch reaktives lymphoides Gewebe (7). Es kann sich als idiopathischer Zustand manifestieren oder mit verschiedenen zugrunde liegenden Faktoren assoziiert sein, wobei Autoimmunerkrankungen wie das Sjögren-Syndrom (25-50% der LIP-Fälle), systemischer Lupus erythematodes (SLE) und rheumatoide Arthritis (RA) die häufigsten Ursachen sind. Immunodefizienzzustände wie HIV und eine gemeinsame variable Immundefizienz (CVID) können ebenfalls mit LIP in Verbindung gebracht werden (8).
Radiologische Anomalien bei LIP zeigen verschiedene Erscheinungsformen, darunter Ground Glass Opazitäten (GGO), zentrilobuläre Noduli und zystische Veränderungen. Ground Glass Opazitäten werden eher im frühen Stadium der Krankheit beschrieben, während Noduli und Zysten bei chronischer, langjähriger LIP vermehrt beobachtet werden (9). Pulmonale Zysten wurden bei 60-80% der Patienten mit LIP festgestellt und zeigten eine durchschnittliche Größe von 16 mm (3-52 mm), bilaterales Auftreten und eine subpleurale oder peri-broncho-vaskuläre Verteilung. Eine zufällige Verteilung ist ebenfalls in der Literatur dokumentiert. Die Lungenfunktionsprüfung zeigt in der Regel ein restriktives Muster mit reduzierter Diffusionskapazität. Eine histologische Bewertung ist unerlässlich, um die Diagnose zu bestätigen.
Die Behandlung von LIP zielt darauf ab, die zugrunde liegende Erkrankung zu behandeln. In Fällen, die mit HIV assoziiert sind, haben antiretrovirale Therapien klinische Wirksamkeit gezeigt und eine Remission der LIP wurde berichtet (10). Für die meisten anderen Fälle ist ein immunsuppressives Regime mit Kortikosteroiden oder eine Kortikosteroid-sparend Therapie zur Verbesserung oder Stabilisierung der Krankheit empfohlen. Eine enge Überwachung mit wiederholten radiologischen Kontrollen wird bei Erkrankungen wie dem Sjögren-Syndrom empfohlen, jedoch ist das optimale Intervall und die Dauer der Nachbeobachtung noch zu bestimmen.
Lymphangioleiomyomatose
Die Lymphangioleiomyomatose (LAM) ist eine seltene zystische Lungenerkrankung, die durch die Proliferation von muskelähnlichen Zellen charakterisiert ist, die Mutationen in den Genen TSC1 (Hamartin) und TSC2 (Tuberin) aufweisen (11). Diese Gene kodieren Proteine, die den Signalweg von Rapamycin (mTOR) regulieren (11). Ein Mangel oder eine Dysfunktion von Hamartin und Tuberin führt zu einer hochregulierten mTOR-Aktivität, was zu einer erhöhten Proteinsynthese und unangemessenen zellulären Proliferation, Migration und Invasion führt. Zusätzliche Effekte von TSC1- und TSC2-Mutationen umfassen die Unterdrückung der Autophagie, einen Wechsel zu glykolytischem Stoffwechsel und die Expression von vaskulären endothelialen Wachstumsfaktoren (=vascular endothelial growth factor/ VEGF-C und VEGF-D) (12). Ein erhöhter Serumspiegel von VEGF-D ist bei 50-70% der Patienten mit LAM messbar und dient als nützlicher diagnostischer und prognostischer Marker (13,14).
Die Rolle von Östrogen in der Pathologie von LAM ist noch nicht vollständig verstanden, jedoch steht eine Progredienz in Verbindung mit hohen Östrogenspiegeln etwa während der Schwangerschaft und Hormonersatztherapie. Ein Pneumothorax während der Schwangerschaft sollte stets den Verdacht auf LAM wecken. Oft wird postmenopausal eine verlangsamte Krankheitsprogression beobachtet. LAM wird entweder als sporadisch oder im Zusammenhang mit dem tuberösen Sklerosekomplex (TSC) klassifiziert. Sporadische LAM zeigt TSC1- und TSC2-Mutationen nur in neoplastischen Läsionen und betrifft hauptsächlich junge Frauen. Im Gegensatz dazu betrifft TSC-assoziierte LAM alle Zellen und tritt bei beiden Geschlechtern auf, wenn auch nicht gleichmässig verteilt (häufiger bei Frauen). TSC-LAM tritt bei 30% der Frauen mit TSC und bei 10-15% der Männer mit TSC auf (15). Das durchschnittliche Alter bei der Diagnose liegt bei 35 Jahren, aber es wurden auch seltene Fälle bei Kindern und älteren Menschen beschrieben.
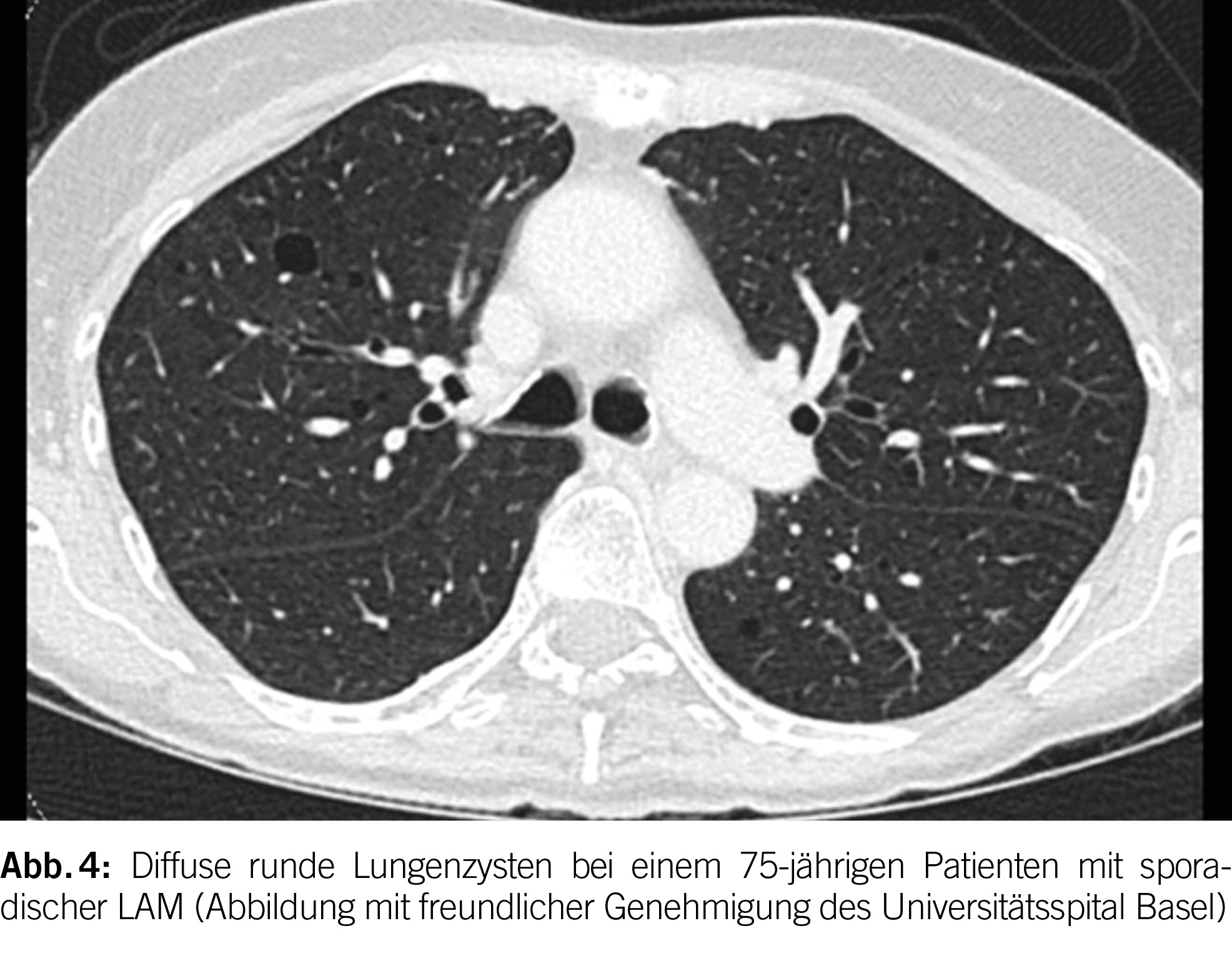
Abb. 5: MRT des Abdomens zeigt ein renalen Angiomyolipom links (orangener Pfeil) bei einer Patientin mit TSC-LAM (Abbildung mit freundlicher Genehmigung des Universitätsspital Basel)
Lungenzysten bei LAM sind typischerweise rund, gleichmäßig in beiden Lungen verteilt und in Form und Grösse relativ einheitlich (Abbildung 4). Bei TSC-LAM treten häufig Lungennoduli mit einer Grösse von 2 bis 14 mm auf, die die zystischen Läsionen begleiten und eine multifokale mikronoduläre Pneumozyten-Hyperplasie repräsentieren. Bei 10% der LAM-Patienten treten weiterhin chylöse Pleuraergüsse auf.
Eine abdominale Beteiligung tritt insbesondere mit renalen Angiomyolipomen auf – gutartigen Tumoren, die ein hohes Blutungsrisiko bei >4 cm Grösse darstellen (Abbildung 5). Abdominale oder mediastinale Lymphangioleiomyome, Aszites und Lymphadenopathien können ebenfalls auftreten.
Der klinische Verlauf der LAM ist durch fortschreitende Dyspnoe bei körperlicher Anstrengung, das wiederholte Auftreten eines Pneumothorax und die Ansammlung von chylöser Flüssigkeit thorakal und abdominal gekennzeichnet. Bei der Lungenfunktion wird häufig ein obstruktiver Defekt und eine Hyperinflation festgestellt. Eine Verschlechterung der Lungenfunktion ist bei Patientinnen im reproduktiven Alter, hohen VEGF-D-Spiegeln und bei Verwendung von östrogenhaltigen Medikamenten festzustellen.
Die Durchführung einer HRCT Untersuchung zum Ausschluss einer LAM ist gerechtfertigt bei Auftreten eines Pneumothorax in der Schwangerschaft oder bei jungen, weiblichen Nichtraucherinnen, bei asymptomatischen Patienten mit TSC sowie bei der zufälligen Entdeckung von Angiomyolipomen oder unerklärlichem chylösem Aszites beziehungsweise Pleuraerguss. Die Leitlinien der ERS (European Respiratory Society) weisen darauf hin, dass die Diagnose von LAM auf der Grundlage charakteristischer zystischer Läsionen bei einem Patienten mit TSC, Angiomyolipom oder Chylothorax gestellt werden kann. Ein VEGF-D-Serumspiegel über 800 pg/ml bei einem Patienten mit typischen Zysten ist ebenfalls diagnostisch für LAM (16).
In der MILES-Studie (17) hat Sirolimus eine Verlangsamung der Progredienz der LAM gezeigt. Evidenzbasierte Empfehlungen schlagen vor, die Sirolimus-Behandlung zu beginnen, wenn das forcierte exspiratorische Volumen in einer Sekunde (FEV1) unter 70% fällt. Die optimale Behandlungsdauer bleibt unklar, und viele Patienten werden unbefristet behandelt. Ein weiterer Aspekt des LAM-Managements umfasst die regelmäßige radiologische Überwachung von Angiomyolipomen. Nachgewiesen ist ein höheres Blutungsrisiko bei Läsionen grösser als 4 cm, wonach sich eine Intervention, wie eine chirurgische Entfernung oder Embolisation, empfiehlt.
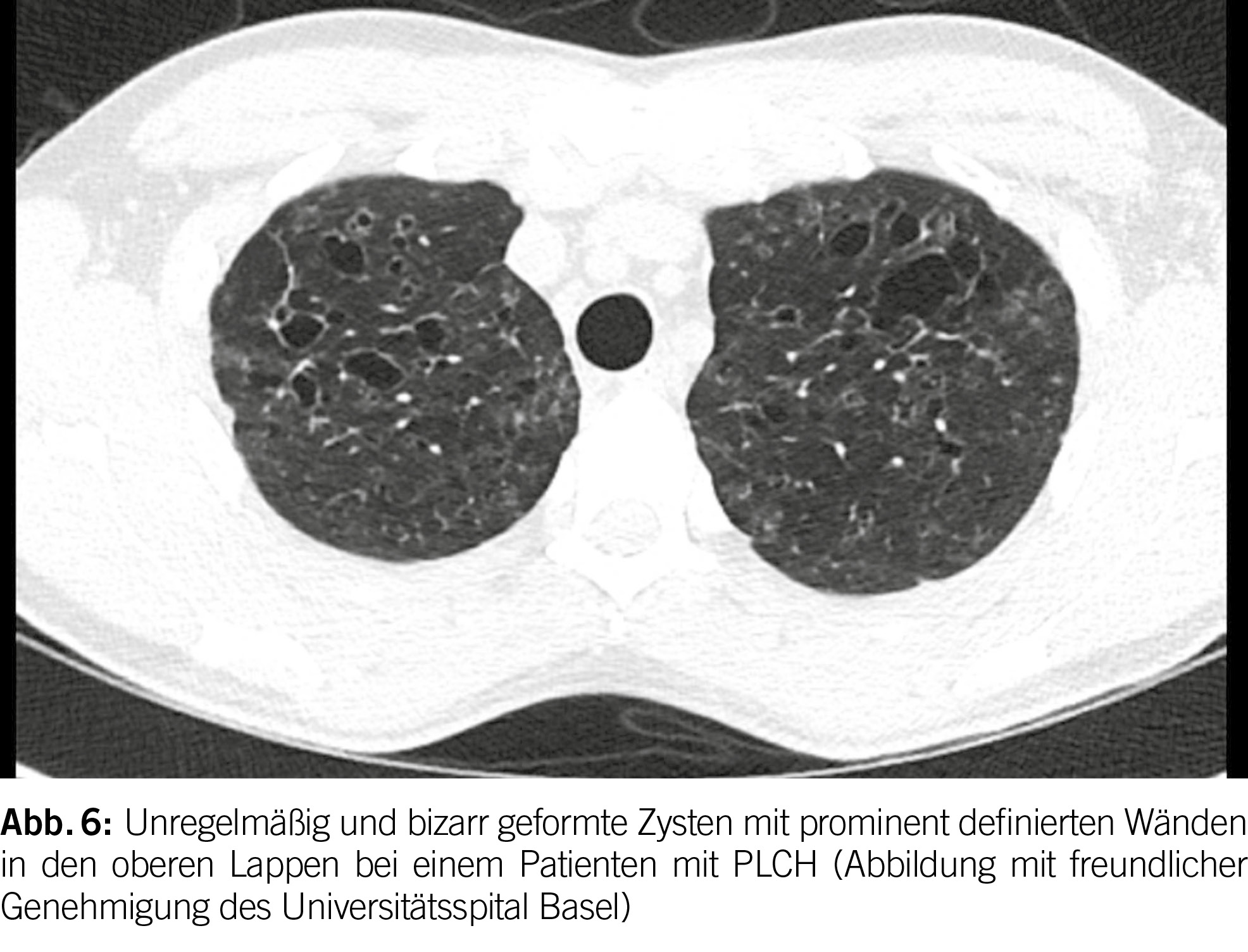
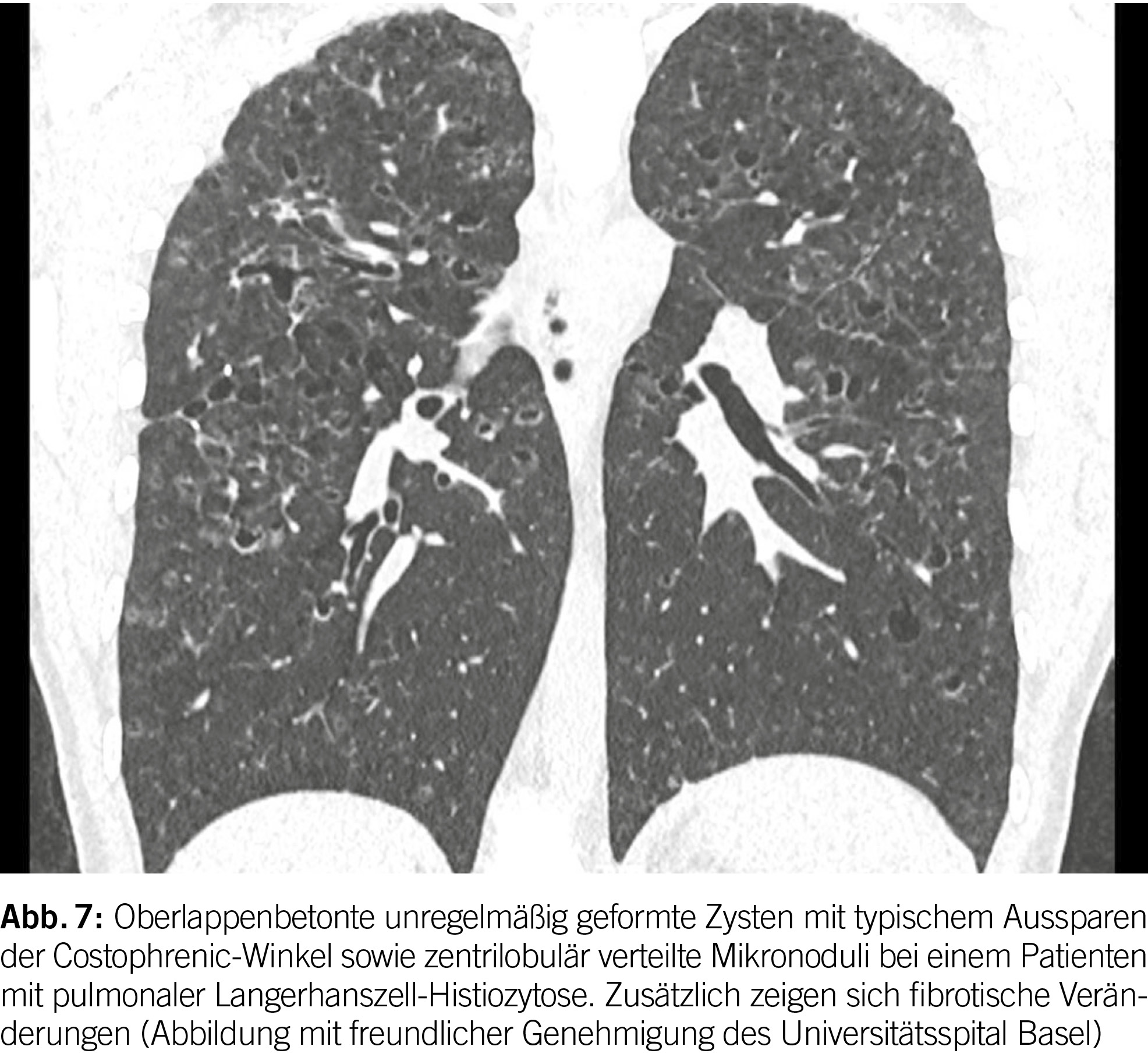
Pulmonale Langerhans-Zell-Histiozytose
Die pulmonale Langerhans-Zell-Histiozytose (PLCH) ist eine systemische Erkrankung, die sich als diffuse zystische interstitielle Lungenerkrankung manifestiert und hauptsächlich junge Erwachsene betrifft, die rauchen. Obwohl die genaue Pathogenese der Krankheit noch diskutiert wird, besteht ein gut dokumentierter Zusammenhang mit Nikotinabusus als begünstigendem Faktor. Die Prävalenz des Zigarettenrauchens liegt bei über 90% der diagnostizierten PLCH-Patienten (18). Somatische Mutationen in den MAPK-Signalwegen, insbesondere BRAF V600E und MAPK2K1, sind in den meisten Fällen nachweisbar (19).
Das klinische Bild ist bei der Mehrheit der Patienten unspezifisch und reicht von minimalen Symptomen bis zu Dyspnoe und Husten. Eine schwerere Präsentation mit konstitutionellen Symptomen wie Gewichtsverlust und Fieber ist ebenfalls möglich, beschränkt sich jedoch auf 20% der Fälle. Etwa 20% der Patienten zeigen extrapulmonale Manifestationen zum Zeitpunkt der Diagnose. Am häufigsten handelt es sich um zystische Knochenläsionen und pathologische Knochenbrüche. Eine skelettale Beteiligung kann pulmonalen Manifestationen vorausgehen und betrifft typischerweise flache Knochen. Eine Hypothalamus-Beteiligung, die zur Entwicklung von Diabetes insipidus führt, tritt bei 5-15% der Patienten auf, während Hautbeteiligung bei weniger als 5% zu beobachten ist. Die Lungenfunktion bleibt bei PLCH in der Regel erhalten. In den frühen Phasen der Krankheit wurde ein restriktives Muster beschrieben, das CT-radiologisch überwiegend nodulären Veränderungen entspricht. In den fortgeschrittenen Phasen der Krankheit zeigt sich lungenfunktionell ein obstruktives Muster, CT-radiologisch einem vorherrschend zystischen Muster entsprechend. Die Diffusionskapazität ist häufig im Verhältnis zu den Veränderungen des Lungenvolumens übermässig reduziert.
Die hochauflösende Computertomographie (HRCT) spielt eine entscheidende Rolle bei der Evaluation von PLCH. Das charakteristische Merkmal ist das Vorhandensein von diffusen Zysten und Noduli, die sich überwiegend in den mittleren bis superioren Bereichen der Lungen ansammeln, wobei der kostophrenische Winkel nicht befallen ist (20) (Abbildung 6). Die Noduli weisen oft eine schlecht definierte Form auf und messen typischerweise zwischen 2 und 10 mm.
Langerhans-Zellen sind spezialisierte dendritische Zellen, die eine entscheidende Rolle bei der Regulation der Schleimhautimmunität spielen (20). In der Pathologie von PLCH umfasst die initiale Läsion die Ansammlung aktivierter Langerhans-Zellen um die terminalen und respiratorischen Bronchiolen. Die Signale, die zur Aktivierung der Langerhans-Zellen führen, sind noch Gegenstand der Debatte, wobei das Rauchen einen prominenten Faktor darstellt (21). Die Langerhans-Zellen und die nachfolgende Rekrutierung von Entzündungszellen tragen zur Bildung von Noduli bei, die der Entwicklung von Remodeling der Atemwege und zystischen Veränderungen vorausgehen (19).
Die Diagnose von PLCH sollte bei jungen Rauchern, die sich mit zystischen und nodulären Infiltraten präsentieren, in Betracht gezogen werden. Eine Anamnese mit aufgetretenem Pneumothorax oder das Vorhandensein von Diabetes insipidus sollte die Aufmerksamkeit der Ärzte auf das mögliche Vorhandensein von PLCH lenken. HRCT ist ein wesentlicher Bestandteil der Untersuchung auf PLCH. Wenn klinische und radiologische Befunde auf PLCH hinweisen, sind weitere invasive diagnostische Verfahren angebracht. Zur Bestätigung der Diagnose kann eine bronchoalveoläre Lavage (BAL) ausreichend sein, wenn mehr als 5% CD1a-positive Zellen identifiziert werden, was eine hohe Spezifität, aber eine geringe Sensitivität zeigt. Weitere invasive diagnostische Methoden wie transbronchiale Biopsie (TBB) mit einer diagnostischen Ausbeute von 30% und Video-assistierte Thorakoskopie (VATS) können erforderlich sein. Ein Fluordesoxyglukose-Positronenemissionstomographie (FDG PET)-Scan kann bei der Diagnose von PLCH hilfreich sein, da Läsionen typischerweise FDG-positiv sind, insbesondere wenn der Verdacht auf extrapulmonale Beteiligung besteht.
Das Management von PLCH konzentriert sich hauptsächlich auf die Raucherentwöhnung und die Behandlung von Komplikationen wie Pneumothorax und respiratorisches Versagen. Eine medikamentöse Therapie mit Immunsuppression wird in der Regel bei Patienten mit beeinträchtigter Lungenfunktion zum Zeitpunkt der Diagnose oder einem Rückgang bei seriellem Testen trotz erfolgreichem Rauchstopp in Betracht gezogen. Die Wahl des Immunsuppressivums bleibt Gegenstand der Debatte, wobei begrenzt verfügbare Daten aus randomisierten kontrollierten Studien vorliegen. Erfahrungen deuten auf eine begrenzte Wirksamkeit von oralen Kortikosteroiden hin, während andere Substanzen wie Azathioprin, Methotrexat und Cladribin eine höhere Wirksamkeit zeigen können. In den letzten Jahren war ein neues Gebiet die targeted therapy, unter Berücksichtigung der hohen Prävalenz von Mutationen in den Genen der MAPK-Signalwege. Vemurafenib, ein BRAF-Kinaseinhibitor, zeigte eine gute Wirksamkeit in der Krankheitskontrolle (22) und eröffnete neue therapeutische Möglichkeiten bei PLCH.
Silviu-Mihail Chirila, silviu-mihail.chirila@usb.ch
Stv. Oberarzt
Universitätsspital Basel
Klinik für Pneumologie
Petersgraben 4
4031 Basel
Interessenskonflikte: Der Autor hat keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
Universitätsspital Basel
Klinik für Pneumologie
Petersgraben 4
4031 Basel
Literatur:
1. Toro JR, Pautler SE, Stewart L, Glenn GM, Weinreich M, Toure O, Wie MH,Schmidt LS, Davis L, Zbar B,et al. Lung cysts, spontaneous pneumothorax, and genetic associations in 89 families with Birt-Hogg-Dube Syndrome. Am J Respir Crit Care Med2007;175:1044–1053
2. Escalon JG, Richards JC, Koelsch T, et al. Isolated Cystic Lung Disease: An Algorithmic Approach to Distinguishing Birt-Hogg-Dubé Syndrome, Lymphangioleiomyomatosis, and Lymphocytic Interstitial Pneumonia. AJR Am J Roentgenol 2019; 212:1260.
3. Zbar B, Alvord WG, Glenn G, Turner M, Pavlovich CP, Schmidt L,Walther M, Choyke P, Weirich G, Hewitt SM,et al. Risk of renal and colonic neoplasms and spontaneous pneumothorax in theBirt-Hogg-Dub ́e syndrome. Cancer Epidemiol Biomarkers Prev2002;11:393–400
4. Toro JR, Wie MH, Glenn GM, et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet 2008; 45:321.
5. Menko FH, van Steensel MA, Giraud S, Friis-Hansen L, Richard S,Ungari S, Nordenskj ̈old M, Hansen TV, Solly J, Maher ER; European BHD Consortium. Birt-Hogg-Dub ́e syndrome: diagnosis and management. Lancet Oncol2009;10:1199–1206
6. Stamatakis L, Metwalli AR, Middelton LA, Marston Linehan W. Diagnosis and management of BHD-associated kidney cancer.Fam Cancer2013;12:397–402
7. Nicholson AG. Lymphocytic interstitial pneumonia and other lymphoproliferative disorders in the lung. Semin Respir Crit CareMed2001;22:409–422.
8. Cha SI, Fessler MB, Cool CD, Schwarz MI, Brown KK. Lymphoid interstitial pneumonia: clinical features, associations and prognosis. Eur Respir J2006;28:364–369.
9. Johkoh T, Müller NL, Pickford HA, et al. Lymphocytic interstitial pneumonia: thin-section CT findings in 22 patients. Radiology 1999; 212:567.
10. Dufour V, Wislez M, Bergot E, Mayaud C, Cadranel J. Improvement of symptomatic human immunodeficiency virus-related lymphoid interstitial pneumonia in patients receiving highly active antiretroviral therapy.Clin Infect Dis2003;36:e127–e130.
11. Tapon N, Ito N, Dickson BJ, Treisman JE, Hariharan IK. The Drosophila tuberous sclerosis complex gene homologs restrict cell growth and cell proliferation. Cell2001;105:345–355
12. Henske EP, McCormack FX. Lymphangioleiomyomatosis – a wolf in sheep’s clothing. J Clin Invest2012;122:3807–3816
13. Young L, Lee HS, Inoue Y, Moss J, Singer LG, Strange C, Nakata K,Barker AF, Chapman JT, Brantly ML,et al.; MILES Trial Group. Serum VEGF-D a concentration as a biomarker of lymphangioleiomyomatosis severity and treatment response: a prospective analysis of the Multicenter International Lymphangioleiomyomatosis Efficacy of Sirolimus (MILES) trial. Lancet Respir Med2013;1:445–452.
14. Young LR, Inoue Y, McCormack FX. Diagnostic potential of serum VEGF-D for lymphangioleiomyomatosis. NEnglJMed2008;358:199–200.
15. Muzykewicz DA, Sharma A, Muse V, Numis AL, Rajagopal J, Thiele EA.TSC1 and TSC2 mutations in patients with lymphangioleiomyomatosis and tuberous sclerosis complex.JMedGenet2009;46:465–468.
16. S. R. Johnson, J. F. Cordier, R. Lazor, V. Cottin, U. Costabel, S. Harari, M. Reynaud-Gaubert, A. Boehler, M. Brauner, H. Popper, F. Bonetti, C. Kingswood, the Review Panel of the ERS LAM Task Force European Respiratory Journal 2010 35: 14-26; DOI: 10.1183/09031936.00076209
17. Francis x. McCormack, Yoshikazu Inoue, Joel Moss, Lianne G. Singer, Charlie Strange, Koh Nakata, Alan F. Barker, Jeffrey T. Chapman, Mark l. Brantly et al., Efficacy and Safety of Sirolimus in Lymphangioleiomyomatosis
18. Vassallo R, Ryu JH, Schroeder DR, Decker PA, Limper AH. Clinical outcomes of pulmonary Langerhans’-cell histiocytosis in adults. N Engl J Med2002;346:484–490
19. Gupta N, Vassallo R, Wikenheiser-Brokamp KA, McCormack FX. Diffuse Cystic Lung Disease. Part II. Am J Respir Crit Care Med 2015; 192:17.
20. Abbott GF, Rosado-de-Christenson ML, Franks TJ, et al. From the archives of the AFIP: pulmonary Langerhans cell histiocytosis. Radiographics 2004; 24:821.
21. Xaubet A, Agust ́ıC, Picado C, Guer ́equiz S, Martos JA, Carri ́on M,Agust ́ı-Vidal A. Bronchoalveolar lavage analysis with anti-T6monoclonal antibody in the evaluation of diffuse lung diseases.Respiration1989;56:161–166
22. Donadieu J, Larabi IA, Tardieu M, Visser J, Hutter C, Sieni E, et al. Vemurafenib for refractory multisystem langerhans cell histiocytosis in children: an international observational study. J Clin Oncol. (2019) 37:2857–65. doi: 10.1200/JCO.19.00456