Einleitung
Interstitielle Pneumopathien (ILD) sind eine Gruppe von heterogenen Lungenparenchym Erkrankungen, welche in 16-35% assoziiert mit einer Konnektivitiden auftreten (CTD-ILD). [1-3] Umgekehrt haben je nach Konnektivitis bis zu 85% der Betroffenen eine subklinische und bis zu 25% eine klinisch manifeste ILD.[4] Bei der Systemischen Sklerose (SSc) ist eine ILD die häufigste Todesursache[5], ähnlich ist das Mortalitätsrisiko bei der Rheumatoiden Arthritis (RA) doppelt so hoch, wenn eine assoziierte ILD besteht.[6] Auch bei der Polymyositis/Dermatomyositis (PM/DM), dem Sjögren Syndrom und der Mixed Connective Tissue Disease (MCTD) muss häufig eine ILD diagnostiziert werden.
Viele Patient/-innen haben zum Zeitpunkt der ILD Detektion bereits eine diagnostizierte Konnektivitis, gelegentlich kann die ILD jedoch als ersten Organbefall manifest werden und extrapulmonale Symptome und Zeichen folgen der ILD Diagnose erst Monate bis Jahre später.[7] So sollten Patient/-innen mit unklassifizierten ILDs jeweils ausgiebig auf eine zugrunde liegende Konnektivitis untersucht werden. Die Therapieoptionen für CTD-ILD haben sich in den letzten Jahren vervielfacht und die Prognose für CTD-ILD Betroffene hat sich bereits verbessert, auch wenn die Evidenz für einige spezifische Konnektivitiden spärlich bleibt.
Dieser Übersichtsartikel hat zum Ziel, die Diagnostik der Konnektivitis assoziierten ILD zu skizzieren, spezifische CTD-ILDs zu besprechen und einen Überblick über die Therapieoptionen zu schaffen.
Abklärungen & Untersuchungen
Anamnese und klinische Untersuchung
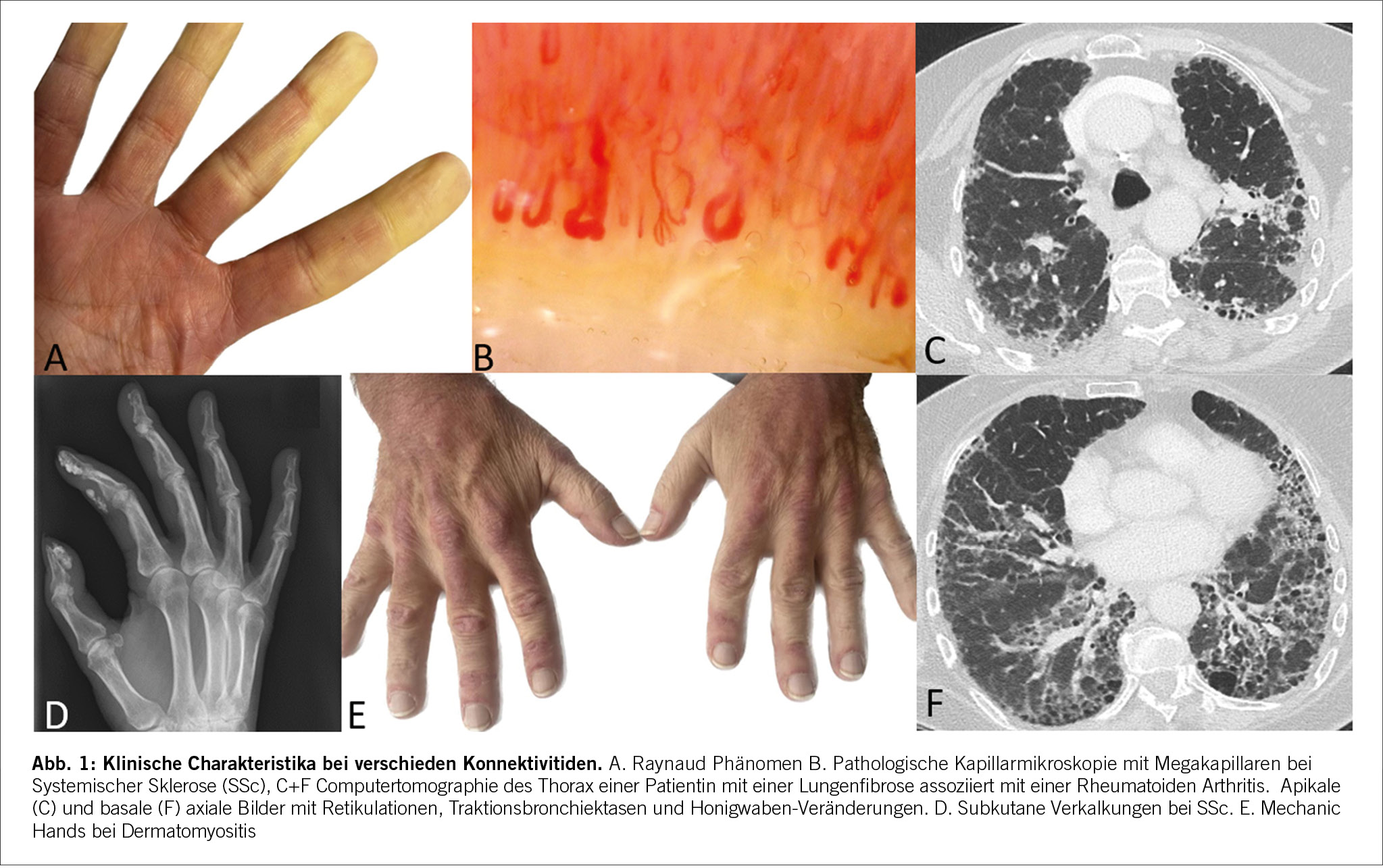
Die Anamnese kann bereits wichtige Hinweise auf das Vorliegen einer Konnektivitis ergeben und ist essenziell, um eine CTD-ILD von ILDs anderer Ursachen zu differenzieren. Neben einer eingehenden Umwelt- und Expositionsanamnese ist die Erfassung von Medikamenten, welche eine ILD verursachen können, gerade im Kontext von CTDs besonders wichtig. Wie bei anderen ILDs, klagen Betroffene oft über eine progrediente Anstrengungs- und später Ruhedyspnoe, trockenen Reizhusten, seltener auch thorakale Schmerzen. Zudem bestehen häufig systemische Beschwerden wie Fatigue, Appetitlosigkeit und ungewollter Gewichtsverlust. Richtungsweisende Konnektivitis-spezifische Symptome sind das Raynaud-Phänomen (Abb. 1), okuläre und orale Sicca-Symptomatik, typische kutane Veränderungen, orale Aphten, proximal betonte Muskelschwäche und Myalgien. Typisch für entzündliche Gelenkbeschwerden sind eine relevante Morgensteifigkeit (mindestens 1 Stunde), Erwachen in der zweiten Nachthälfte und Verbesserung bei Bewegung.
Die klinische Untersuchung spielt bei der Zuordnung der möglichen Grunderkrankung eine wichtige Rolle und beinhaltet einen kompletten (internistischen) Status. Lungenauskultatorisch variieren die Befunde je nach Schweregrad der ILD. Typisch ist ein basal betontes inspiratorisches Knisterrasseln (Sklerosiphonie). Kutan kann bei der SSc und auch bei der MCTD eine Hautfibrose bestehen, diese findet sich typischerweise betont an den Extremitäten und ist begleitet von Sklerodaktylie, möglicherweise in Kombination mit Fingerkuppennekrosen und aktiven Ulzera. Häufig finden sich Teleangektasien, ebenfalls bevorzugt an den Händen und im Gesicht. Für die Dermatomyositis sind unter anderem Gottron Papeln, Mechanic Hands, und der heliotrope Rush charakteristisch. Alle Konnektivitiden können sich mit einem artikulären Befall manifestieren. Hinweise auf entzündliche Veränderungen wie Arthritiden oder Tendovaginitiden sind Schwellung, Rötung und Druckdolenz mit positivem Gänslenzeichen. Eine verminderte Muskelkraft findet sich häufig bei der DM/PM, kann aber auch bei allen anderen Konnektivitiden mit Muskelbeteiligung vorliegen. Die proximale Muskulatur ist häufig betroffen, wobei der Schweregrad sehr unterschiedlich ist. Mittels isometrischer Muskelkraftmessung können einzelne Muskelgruppen separat geprüft werden und so können der Schweregrad einer Muskelbeteiligung objektiviert und der Verlauf und das Therapieansprechen beurteilt werden. Bei der Palpation des Halses können zudem geschwollene Speicheldrüsen oder Lymphknoten getastet werden.
Lungenfunktion und körperliche Leistungstests
Die Lungenfunktionsprüfung eignet sich zur vielseitigen Abklärung von Dyspnoe, Husten und thorakalen Beschwerden und spielt in der Diagnose und Verlaufsbeurteilung von ILDs eine zentrale Rolle. Fibrotische ILDs zeigen sich typischerweise mit einer Restriktion und einer Einschränkung der Diffusionskapazität für CO (DLCO). Die Spirometrie kann eine Obstruktion zeigen oder Hinweise auf eine Restriktion ergeben. Zum Beweis oder Ausschluss einer Restriktion braucht es jedoch eine Bodyplethysmographie, welche neben den dynamischen auch die statischen Lungenvolumina messen kann. Die Restriktion ist definiert als eine Totale Lungenkapazität (TLC) < 5. Perzentile.[8] Neben einer ILD kann auch eine muskuläre Einschränkung, wie sie bei Konnektivitiden mit generalisierten Myopathien (DM/PM) oder schwerer Sarkopenie vorkommt, eine Restriktion verursachen. Die Diffusionskapazität für CO widerspiegelt die Gasaustauschfähigkeit des kardiopulmonalen Systems und ist häufig bei ILDs eingeschränkt. Patient/-innen mit Konnektivitiden haben ein hohes Risiko einer Pulmonalen Hypertonie (PH) und bei eingeschränkter DLCO sollte auch an die PH als Ursache einer Diffusionsstörung gedacht werden, insbesondere wenn die DLCO im Verhältnis zur Vitalkapazität überproportional tief ist (reduzierte KCO). Die Forcierte Vitalkapazität (FVC) und die DLCO sind die relevanten Verlaufsparameter, um eine Progression der ILD und das Therapieansprechen zu beurteilen. Der 6-Minuten-Gehtest liefert wertvolle Informationen zur körperlichen Leistungsfähigkeit und kann eine Belastungshypoxämie dokumentieren. Einerseits kann die 6-Minuten-Gehstrecke (6MWD) als Verlaufsparameter genutzt werden, andererseits eine Sauerstofftherapie bedarfsgerecht verordnet werden. Einschränkungen der 6MWD sind unspezifisch und neben der ILD kann eine PH oder eine muskuloskelettale Limitation die 6MWD reduzieren. Mittels einer Spiroergometrie können eine Dyspnoe oder körperliche Leistungsintoleranz besser differenziert werden. Zudem ist die Untersuchung sensitiver als der Gehtest und sehr gut, um Leistungseinschränkungen auch bei jungen und fitten Patient/-innen nachzuweisen. Die Spiroergometrie kann eine restriktive Ventilationsstörung von einer PH, einer Kardiopathie oder einer muskulären Limitation differenzieren.
Radiologie und Pathologie
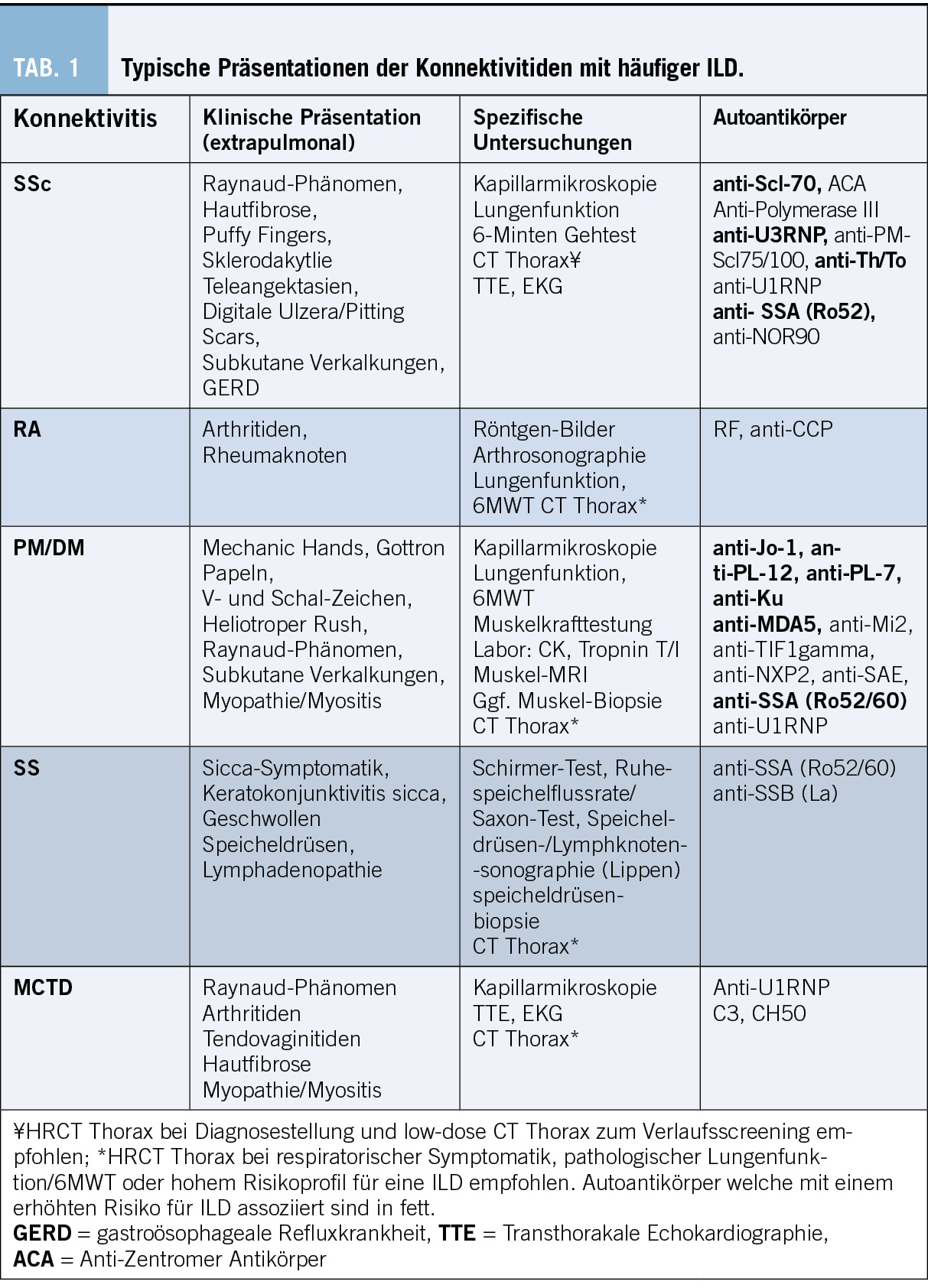
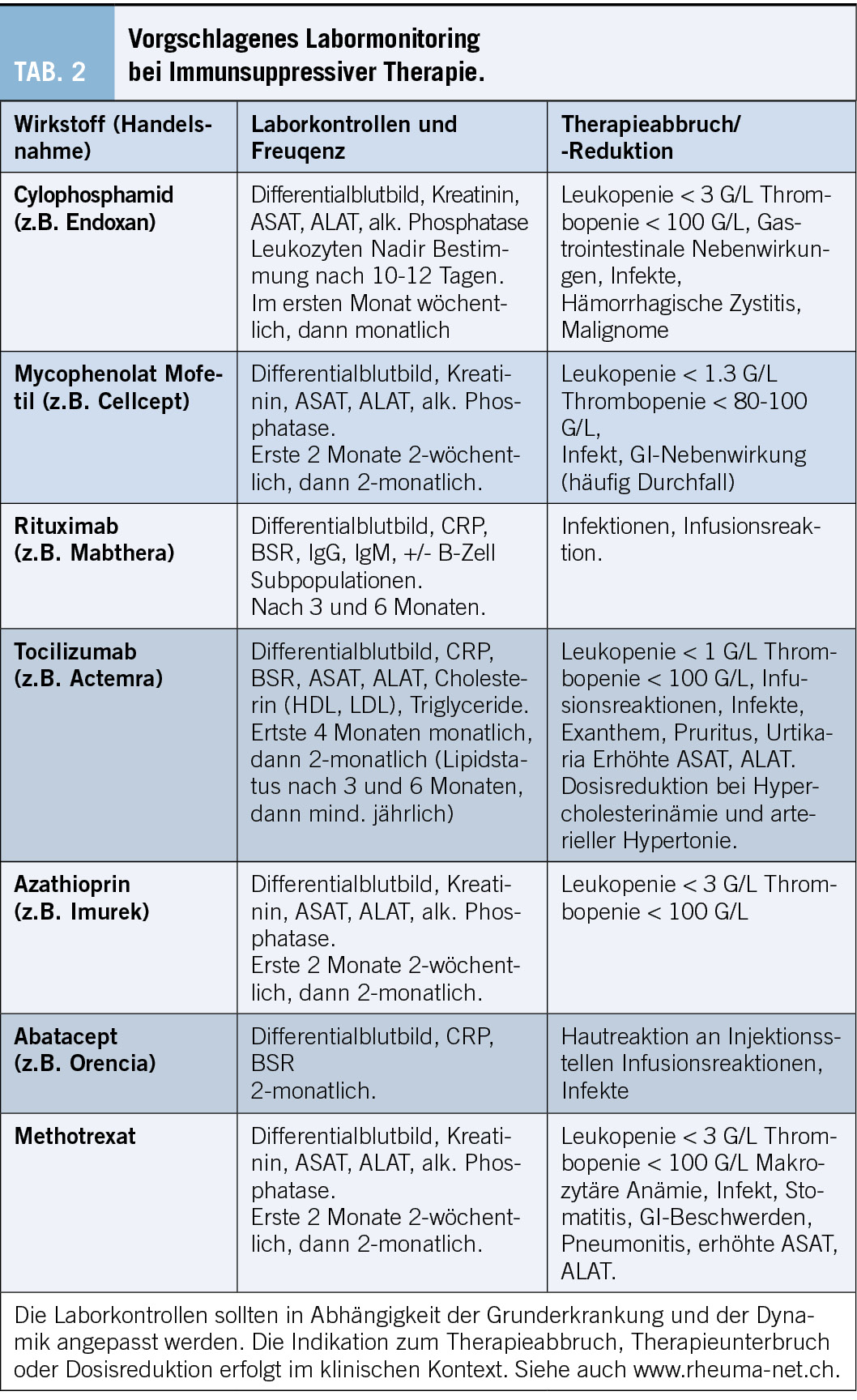
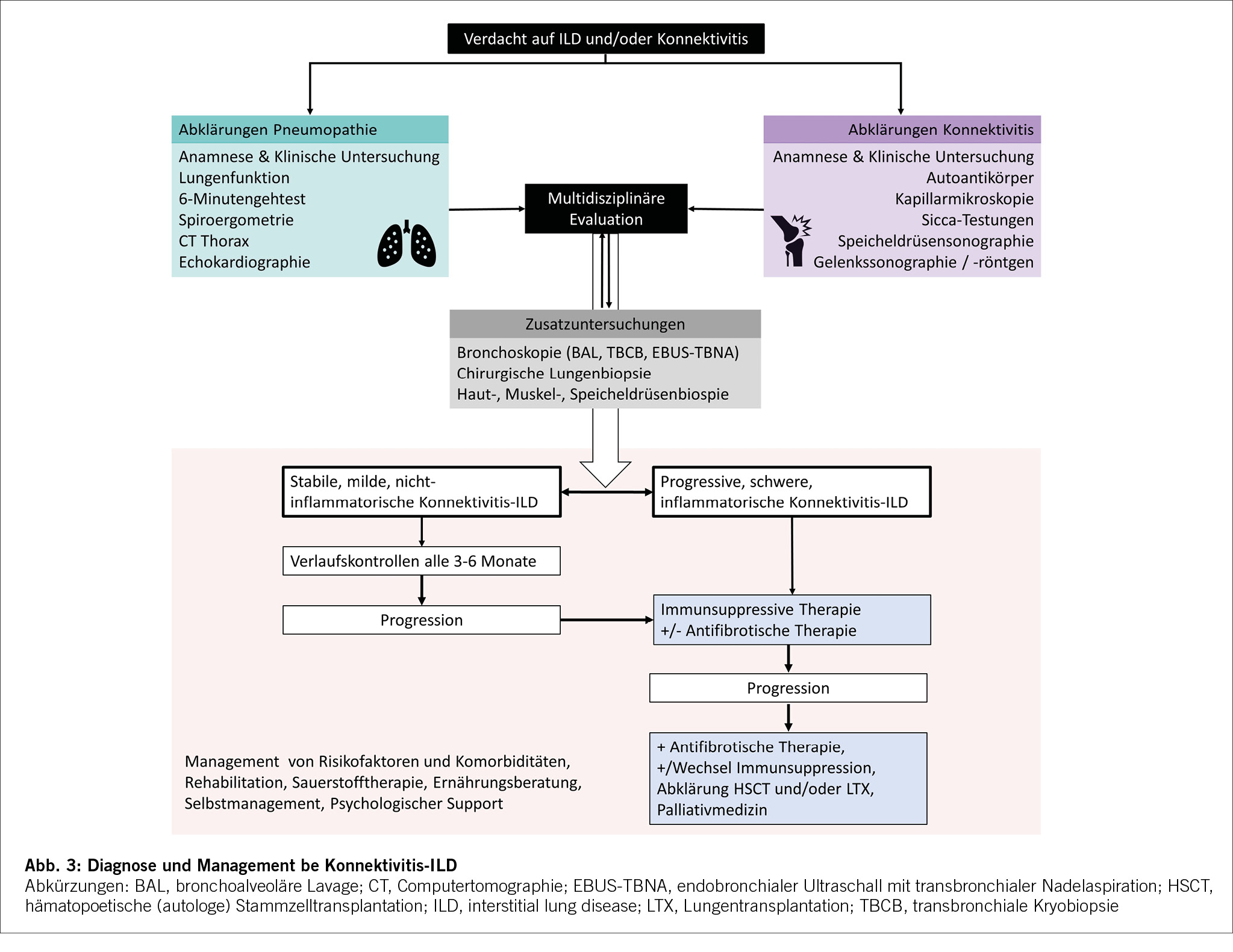
Die Bildgebung ist für die Diagnose, Differenzierung und Verlaufsbeurteilung bei ILD von zentraler Bedeutung und eine CTD-ILD kann nur mittels hochauflösender Computertomographie (HRCT) diagnostiziert werden.[9],[10] Konventionelle Thorax Röntgenbilder können zwar ein verringertes Lungenvolumen und retikuläre oder noduläre Transparenzminderungen zeigen, diese sind aber unspezifisch und erlauben keine definitive ILD Diagnose. Auch ist das Röntgen nicht sensitiv genug, um eine ILD ausschliessen zu können. Im CT des Thorax kann neben der ILD-Diagnose, auch das spezifische Muster (Abb. 2) und der Schweregrad der ILD dargestellt werden. Der prozentuale Anteil der Lunge, welcher verändert ist, sollte gerade bei CTD-ILD auch quantitativ angegeben werden, was therapeutische Entscheidungen und die Verlaufsbeurteilung erleichtert. Die am häufigsten auftretenden Muster sind die non-specific interstitial pneumonia (NSIP) welche häufig bei der Systemischen Sklerose zu finden ist und die usual interstitial pneumonia (UIP) welche neben der NSIP bei der Rheumatoiden Arthritis häufig ist. Auch Organisierende Pneumonien (OP) kommen bei Konnektivitiden häufig zur Darstellung. Die Lymphozytäre Interstitielle Pneumopathie (LIP), welche sich mit groundglass Opazitäten, Retikulationen und dünnwandigen Zysten präsentiert, ist zwar selten, aber charakteristisch für das Sjögren Syndrom. Neben einer ILD kann das CT auch Bronchopathien (Bronchiektasen, Bronchiolitis), Pleuraergüsse, intrapulmonale Rheumaknoten oder indirekte Zeichen für eine PH wie z.B. verbreiterte Pulmonalarterien oder ein vergrösserter rechter Ventrikel identifizieren. Angesichts des erhöhten Malignomrisikos bei Entzündungserkrankungen können regelmässige Thorax CTs gerade bei (ehemaligen) Rauchern auch dem Tumorscreening dienen.[11-12] Bei Betroffenen von Systemischer Sklerose wird empfohlen, insbesondere in den ersten Krankheitsjahren, jährlich eine neu auftretende oder progrediente ILD zu suchen auch bei asymptomatischen Personen. Das jährliche Screening kann mittels low-dose CT Thorax durchgeführt werden, um die Strahlenbelastung minimal zu halten. Im Abklärungsprozess der ILDs sollte die Indikation einer chirurgischen Lungenbiopsie oder einer Bronchoskopie mit bronchoalveolärer Lavage (BAL), Ultraschall gesteuerter Punktionen der thorakalen Lymphknoten (EBUS-TBNA) oder transbronchiale Kryobiopsien des Lungenparenchyms anlässlich einer Multidisziplinären Diskussion (MDD) gestellt werden.[13-14] Gerade wenn bei einer neu diagnostizierten ILD anamnestisch oder klinisch der Verdacht auf eine Konnektivitis besteht, lohnt es sich zuerst die nicht-invasive Diagnostik zu komplettieren, bevor eine Lungenbiopsie geplant wird. Insgesamt sollte der diagnostische Nutzen gegen das peri-interventionelle Risiko abgewogen werden. Bei bekannter Konnektivitis ist in den meisten Fällen weder eine Bronchoskopie noch eine chirurgische Lungenbiopsie zur Abklärung der ILD nötig. Eine BAL kann jedoch vor Beginn einer immunsuppressiven Therapie indiziert sein, um eine infektiöse Exazerbation einer ILD ausschliessen.[15] Bei hilärer oder mediastinaler Lymphadenopathie kann eine EBUS-TBNA der Lymphknoten hilfreich sein, um neben dem Infekt auch ein Lymphom auszuschliessen. An letzteres ist aufgrund der erhöhten Inzidenz insbesondere beim Sjögren Syndrom zu denken.[16] Laboruntersuchungen und Autoantikörper Laboranalytisch kann ein inflammatorisches Syndrom mit erhöhtem CRP und Blutsenkungsgeschwindigkeit bestehen. Beispielsweise präsentieren bei der SSc 20-35 % ein persistierend inflammatorisches Syndrom, welches mit einer pulmonalen Manifestation und höheren Mortalität assoziiert ist [17-18]. Die Immunglobuline (IgG, IgA, IgM), Immunelektrophorese und die Komplementfaktoren (C3, C4) können bei Konnektivitiden abnormal und hinweisend auf eine Krankheitsaktivität sein, zeigen sich aber häufig wie auch das CRP normal. Die CK und das Troponin T sind bei der PM/DM meistens erhöht, wobei ein normaler Wert eine Myositis nicht ausschliesst. Bei Verdacht auf eine kardiale Beteiligung sollte das Troponin I bestimmt werden, welches im Vergleich zu Troponin T spezifisch für den Herzmuskel ist. Bei der Myositis mit hohen Muskelenzymen zeigt sich meistens auch ein Anstieg der Transaminasen, als Ausdruck der Muskelschädigung. Die Antinuklearen Antikörper (ANA), welche mittels indirekter Immunfluoreszenzassay (IFA) bestimmt werden, dienen als Screening für Konnektivitiden. Bei einem erhöhten Titer werden dazu ein oder mehrere Muster beschrieben (homogen, (fein-)granulär, nukleolär, zentromer oder zytoplasmatisch). [19] In Abhängigkeit des Musters und des klinischen Kontexts erfolgt die Bestimmung der spezifischen Subtypen (Tab. 1). Zu beachten ist, dass die zytoplasmatischen Antikörper, hauptsächlich Myositis-assoziierte Antikörper, nicht unbedingt mit einem erhöhten ANA-Titer angezeigt werden. Je nach Labor werden diese separat angegeben, weshalb bei klinischem Verdacht auf eine PM/DM auch bei normalem ANA Titer die Bestimmung der Myositis-Antikörper erfolgen sollte. Die Präsenz von bestimmten Autoantikörpern (anti-Polymerase III bei SSc und anti-TIF1ƴd, anti-NXP2 und anti-SAE bei DM/PM) ist mit einem erhöhten Risiko einer Neoplasie assoziiert. In solchen Fällen ist ein Tumorscreening empfohlen.[20] In rheumatologischen Kohorten sind die Sensitivität und Spezifität eines positiven Rheumafaktors (RF) ca. 72% und 80% und die Sensitivität und Spezifität des anti – cyclic citrullinated peptide (CCP) ca. 66% und 90% für die Diagnose einer RA.[21] Eine Seropositivität für RF oder anti-CCP ist ein Risikofaktor für RA-assoziierte ILD.[22] Spezifische extrapulmonale Untersuchungen Röntgen-Bilder der Gelenke können (post-)entzündliche Erosionen darstellen. Diese finden sich häufig an Händen und Füssen und können durch suggestive Lokalisationen oder Morphologien hinweisend auf eine Konnektivitis sein. Auch subkutane Verkalkungen, wie sie bei der SSc und DM/PM beobachtet werden, lassen sich radiologisch gut darstellen (Abb. 2). Röntgen-Bilder helfen aber auch differentialdiagnostisch eine Kristallarthopathie oder degenerative Veränderungen zu unterscheiden.[23] Die Arthrosonographie ist in der Diagnostik bei Gelenksbeschwerden eine sehr verbreitete Untersuchungsmethode. Durch die immer besser werdende Auflösung und Technik lassen sich mit dem Ultraschall, durch Darstellung von Erguss, Hyperämie und anderen pathologischen Veränderungen, entzündliche von degenerativen Gelenkserkrankungen gut unterscheiden [24]. Ähnlich wie beim Röntgen können entzündliche Veränderungen im Rahmen unterschiedlicher Grunderkrankungen voneinander unterschieden werden. Auch dient der Ultraschall, insbesondere bei der RA zur Einschätzung des Grades der Krankheitsaktivität und somit auch des therapeutischen Ansprechens. Weichteilveränderungen wie Rheumaknoten oder subkutane Verkalkungen können sonographisch ebenfalls dargestellt werden. Zur Abklärung eines unklaren Gelenksergusses sollte eine Gelenkspunktion erfolgen. Diese ist die zuverlässigste Methode, um zwischen einem entzündlichen und nicht-entzündlichen Erguss zu unterscheiden. Eine Zellzahl unter 2000/μl spricht gegen eine entzündliche Gelenksproblematik. Hier kann mit direktem Nachweis von Kristallen differenzialdiagnostisch eine Kristallarthropathie unterschieden werden.[25] Die Kapillarmikroskopie ist eine nicht-invasive Methode, um die Kapillaren im Nagelbett der Finger darzustellen (Abb. 2). Sie spielt bei der Diagnostik von Konnektivitiden eine wichtige Rolle, insbesondere bei der SSc, wo sie auch in den Klassifikationskriterien berücksichtigt wird.[26] Pathologische Veränderungen, wie Megkapillaren oder eine verminderte Kapillardichte sind sehr suggestiv für eine zugrundeliegende Konnektitivs.[27] Solche Veränderungen können bereits sehr früh im Krankheitsverlauf beobachtet werden, noch bevor ausser dem Raynaud-Phänomen andere Beschwerden bestehen. Bei der SSc werden verschiedene Muster unterschieden (early-, active, und late-pattern nach Cutolo). In einer retrospektiven Studie wurde eine Korrelation zwischen Lungenvolumina und der Kapillardichte beschrieben mit einem höheren Risiko für eine ILD bei pathologisch verminderter Kapillardichte.[28] Die Sicca-Testung mittels Schirmer Test (Tränenproduktion) und Bestimmung der Ruhespeichelflussrate (oder Saxon-Test) sind einfache Methoden, um ein Sicca-Syndrom zu objektivieren. Eine Sicca-Testung sollte bei allen Betroffenen mit der Verdachtsdiagnose eines Sjögren-Syndroms erfolgen, auch wenn keine subjektive Sicca-Symptomatik vorliegt. Aufgrund der schlechten Korrelation mit den angegebenen Symptomen muss die Sicca-Testung immer im Gesamtkontext interpretiert werden. [29] Die Speicheldrüsensonographie ist eine weitere nicht-invasive Methode, um typische Veränderungen bei einem Sjögren-Syndrom darzustellen. Die obengenannten Untersuchungen werden als Puzzlesteine zu einer Konnektivitis und ILD Diagnose zusammengetragen (Abb. 3). Eine interdisziplinäre Beurteilung oder Besprechung an welcher Spezialist/-innen aus der Pneumologie, Rheumatologie, Radiologie und falls nötig Pathologie teilnehmen, kann die diagnostische Sicherheit und Koordination der Behandlung deutlich vereinfachen und verbessern. [13-14]
Spezifische Konnektivitiden
Die häufigsten Konnektivitiden mit assoziierter ILD sind die Systemische Sklerose, die Rheumatoide Arthritis, die Myositiden, und das Sjögren Syndrom und die Mixed Connective Tissue Disease (MCTD). Seltener können ILDs auch beim Systemischen Lupus Erythematodes und bei ANCA-assoziierten Kleingefässvaskulitiden auftreten.

Systemische Sklerose
Die Systemische Sklerose ist durch Vaskulopathie, Inflammation und Fibrose verschiedener innerer Organe charakterisiert. Klinisch wird häufig zwischen dem diffus kutanen und dem limitiert kutanen Phänotyp unterschieden. Die Lungenfibrose (SSc-ILD) ist mit einer Prävalenz von 50-80% eine häufige Organmanifestation, und bis zu 30% der Betroffenen mit SSc-ILD entwickeln eine progressive ILD mit zunehmender Leistungsintoleranz, relevantem Abfall der Vitalkapazität in der Lungenfunktion und Progression der ILD im CT [30-33]. Da über die Hälfte der Patient/-innen in den ersten 3 Jahren nach Diagnosestellung eine SSc-ILD entwickeln,[34] sind regelmässige Screenings und Verlaufskontrolle bei allen Betroffenen sehr wichtig, insbesondere in den ersten Jahren. Ein höheres Risiko eine ILD zu entwickelt findet sich bei Männern, dem diffus kutanen Phänotyp, afroamerikanischer Herkunft und bei Nachweis von bestimmen Antikörpern, wie anti-Scl70, anti-Th/To und anti-U3 RNP (Tab. 1). Allerdings ist zu beachten, dass alle SSc Betroffenen eine SSc-ILD entwickeln können und entgegen früheren Vorstellungen ca. 20% der SSc-ILD Patient/-innen einen limitiert kutanen Phänotyp mit positiven Anticentromer- und negativen anti-Scl70 Antikörpern haben.[32-33] Risikofaktoren für eine SSc-ILD Progression sind die Präsenz von anti-Scl70 Antikörper, höheres Alter bei Diagnose, ethnische afroamerikanische Herkunft, eine bereits tiefe FVC oder DLCO, sowie eine extensive Fibrose im CT bei Diagnose der SSc-ILD. [35] Mehr als >80% der SSc Betroffenen leiden an einer ösophageale Dysfunktion mit Reflux, Dysphagie und Regurgitation. [36] Ein kausaler Zusammenhang zwischen ösophagealer Dysfunktion und SSc-ILD wird vermutet, konnte jedoch bisher nicht bewiesen werden[37], allerdings zeigt eine rezente Studie eine Korrelation zwischen Schweregrad der Motilitätsstörung und verminderter DLCO. [38]
Rheumatoide Arthritis
Eine ILD kann bei 30-60% der Patient/-innen mit Rheumatoider Arthritis (RA) gefunden werden, wobei eine symptomatische oder prognostisch relevante ILD nur in 10-30% der RA Fällen besteht.[22],[39] Folglich ist die ILD ist bei RA relativ gesehen zwar etwas seltener als bei der Systemischen Sklerose, da die RA aber ca. 1% der nordeuropäischen Bevölkerung betrifft,[40] begegnet man der RA-ILD in der Praxis nicht so selten. Eine dänische Studie berichtet, dass die 5-Jahres-Mortalität bei RA doppelt so hoch ist, wenn eine assoziierte ILD besteht (36% versus 18%). Dies betont die Wichtigkeit einer engmaschigen Betreuung und optimalen Therapie der Patient/-innen mit RA-ILD.[41-42]
Die RA ist etwa dreimal häufiger bei Frauen als bei Männern,[40] das Risko für eine RA-ILD bei Männern jedoch höher.[39] Die Ursache hierfür ist nicht ganz klar, wobei das bei Männern häufigere Zigarettenrauchen als klarer Risikofaktor für eine ILD bei RA angesehen wird.[43] Bei genetischer Veranlagung und bestimmten Umweltfaktoren (z.B. Rauchen) kann es zu einer Citrullinierung von Proteinen (posttranslationale Umwandlung von Arginin zu Citrullin), Bildung von Antikörpern (RF, anti-CCP) und einer Immunreaktion kommen. Eine Hypothese besagt, dass der Ursprung der Immunreaktion im synovialen Gewebe liegt und es auf Grund von ähnlichen Antigenen in der Lunge sekundär zu einer Immunreaktion in der Lunge kommt. Dies wird durch die klinische Beobachtung gestützt, dass die RA-ILD der artikulären RA-Manifestation meist nachfolgt. Gelegentlich ist die ILD jedoch die erste Manifestation der RA, hierzu passt die Hypothese eines initialen Triggers in mukosalen Geweben (oral, pulmonal, gastroinstestinal) mit folgender Transition in die synovialen Gelenke.[44]
Das häufigste radiologische Muster der RA-ILD ist die UIP, welche vor allem bei älteren männlichen Rauchern häufig und ein prognostisch ungünstiger Faktor ist.[43] Des Weiteren findet sich im CT des Thorax häufig eine NSIP, OP, sowie Bronchiektasen, intrapulmonale Rheumaknoten oder ein Pleuraerguss als Zeichen einer (Poly-)Serositis (Tab. 1). Der RA-ILD Verlauf ist heterogen und schwierig vorauszusagen, während viele Betroffene, insbesondere solche mit einer milden RA-ILD, über Jahre eine stabile Lungenfunktion haben, gibt es solche, welche eine rasche Progression zeigen, welche mit einer progredienten Einschränkung der körperlichen Leistungsfähigkeit, der Lebensqualität und einer hohen Mortalität assoziiert ist.[45]
Dermatomyositis/Polymyositis/Anti-Synthetase Syndrom
Global haben etwa 40% der Patient/-innen mit PM/DM eine assoziierte ILD, wobei die ILD in dieser Population mit 50% in Asien viel häufiger ist als mit 26% in Europa.[46] Anti-MDA5 Antikörper sind mit einem rasch progressiven Verlauf der ILD assoziiert, während anti- Jo1, anti-PL 7/12 und anti-KU eher chronisch progressiv verlaufen und eine bessere Prognose aufweisen (Tab. 1).[20] Eine schwere und generalisierte Muskelbeteiligung kann zusätzlich zu einer Insuffizienz der Atemmuskulatur mit verminderter Atemmuskelkraft, einer extrapulmonalen Restriktion und potentiellen Hypoventilation mit Hyperkapnie führen.[47]
Bei der Diagnose einer DM/PM in höherem Alter sollte auch ein Tumorscreening erfolgen, da das Risiko einer Neoplasie auch in Abhängigkeit von gewissen Antikörpern hier deutlich erhöht ist.
Sjögren Syndrom
Das Sjögren-Syndrom ist durch entzündliche Veränderungen der Speicheldrüsen und Tränendrüsen charakterisiert. Als extraglanduläre Manifestation zeigt sich beim Sjögren-Syndrom in ca. 20% der Fälle eine symptomatische ILD. Wie bei allen Konnektivitiden kann die assoziierte ILD der Diagnose des Sjögren-Syndroms um Jahre vorausgehen. [48][49] Radiologisch sind die LIP und Follikuläre Bronchiolitis charakteristisch für das Sjögren-Syndrom machen aber insgesamt weniger als 10% der ILD-Muster aus, häufiger sind eine NSIP, UIP oder OP zu beobachten.
Risikofaktoren für einen schweren Verlauf und erhöhte Mortalität bei Sjögren Syndrom assoziierter ILD sind verminderte dynamische Lungenvolumina und eine ausgeprägte radiologische Fibrose bei Diagnosestellung.[50] Wenn Lungenbiopsien durchgeführt werden müssen, wird eine hohe Dichte an Fibroblastenfoci als schlechtes prognostisches Zeichen gewertet werden.[51]
Eine weitere häufige pulmonale Manifestation bei Sjögren Syndrom ist der tracheobronchiale Befall, welcher einerseits durch den glandulären Befall mit Sicca-Problematik, andererseits durch eine direkte lymphozytäre Infiltration der tracheobronchialen Schleimhaut bedingt sein kann. Die Betroffenen können folglich an einem sehr einschränkenden Husten leiden. [52]
Mixed Connective Tissue Disease
Die MCTD ist eine seltene Erkrankung, welche aber in über 50% der Fälle mit einer ILD einhergeht.[53] Klinisch präsentieren die Betroffenen oft ein Raynaud Phänomen, Puffy Fingers, Arthritiden, Polyserositiden, Myositis oder Ösophagus Dysmotilität. Die Präsenz der anti-U1RNP Antikörper für die Diagnosestellung ist obligat. [54]
Interstitial Pneumonia with Autoimmune Features
Der Begriff Interstitial Pneumonia with Autoimmune Features (IPAF) wurde 2015 geschaffen, um die Nomenklatur für idiopathische ILDs, bei welchen sich Zeichen einer Konnektivitis und Autoimmunität zeigen, zu vereinheitlichen. Es handelt sich um Fälle, welche auch nach der multidisziplinären Diskussion am ILD-Board (MDD) unklassifiziert bleiben. Die rheumatologische Aufarbeitung kann keine Konnektivitis diagnostizieren, findet aber klinisches Zeichen (z.B. Raynaud Phänomen, inflammatorische Arthritis) und einen positiven Antikörper im Serum (z.B. ANA ≥1:320, RF ≥ 2x der Norm). Zudem wird ein «inflammatorisches» radiologisches oder pathologisches ILD-Muster gefordert (z.B. NSIP, OP).[55-57] IPAF ist bisher keine ILD-Diagnose, sondern eine Terminologie für die klinische Forschung, welche hoffentlich ein besseres Verständnis für diesen Phänotyp liefern wird. Der IPAF-Begriff kann aber die Kommunikation und Therapieentscheidungen für ILD-Spezialisten vereinfachen.
Wahrscheinlich auf Grund der Heterogenität in der IPAF-Gruppe liegt die Prognose zwischen der rasch progredienten IPF und der öfters stabilen Konnektivitis-ILDs.[56],[58] Das optimale Management für diese Patient/-innen aktuell noch nicht ganz klar, wichtig ist sicherlich eine interdisziplinäre Beurteilung und regelmässige Reevaluation, da sich oft im Laufe der Zeit weitere klinische oder laboranalytische Hinweise auf eine spezifische ILD ergeben, und auch die IPAF progressiv verlaufen kann.[59],[45]
Differentialdiagnosen
Eine wichtige Differenzialdiagnose zur CTD-ILD ist die Idiopathische Lungenfibrose (IPF), für welche wie bei der RA-ILD das UIP-Muster im HRCT typisch ist. Im Gegensatz zur CTD-ILD verläuft die IPF immer progredient und ist mit einer hohen Mortalität assoziiert. Auch die Therapie (ausschliesslich Antifibrotika) unterscheidet sich signifikant von der Behandlung der CTD-ILD, was die Differenzierung essenziell macht.[60] Gerade radiologisch kann sich eine fibrotische Hypersensitivitätspneumonitis (HP) ähnlich wie eine CTD-ILD zeigen, hier ist eine gründliche Expositionsanamnese und Falldiskussion am ILD-Board wichtig.[13-14] Medikamentös-toxische ILDs treten gelegentlich im Kontext der CTD Therapien auf, wobei bei einer Vielzahl von Immunmodulatoren eine Pneumotoxizität beschrieben wird.[88] Das gefürchtete Methotrexat (MTX) verursacht in seltenen Fällen schwere subakute bis akute Pneumonitiden die fatal verlaufen können.[61] Typischerweise treten die Pneumonitiden in den ersten Behandlungsmonaten auf und eine Kausalität zwischen der Therapie und der ILD-Exacerbation ist nicht immer einfach zu evaluieren.[62]
Therapie
Medikamentöse Therapie
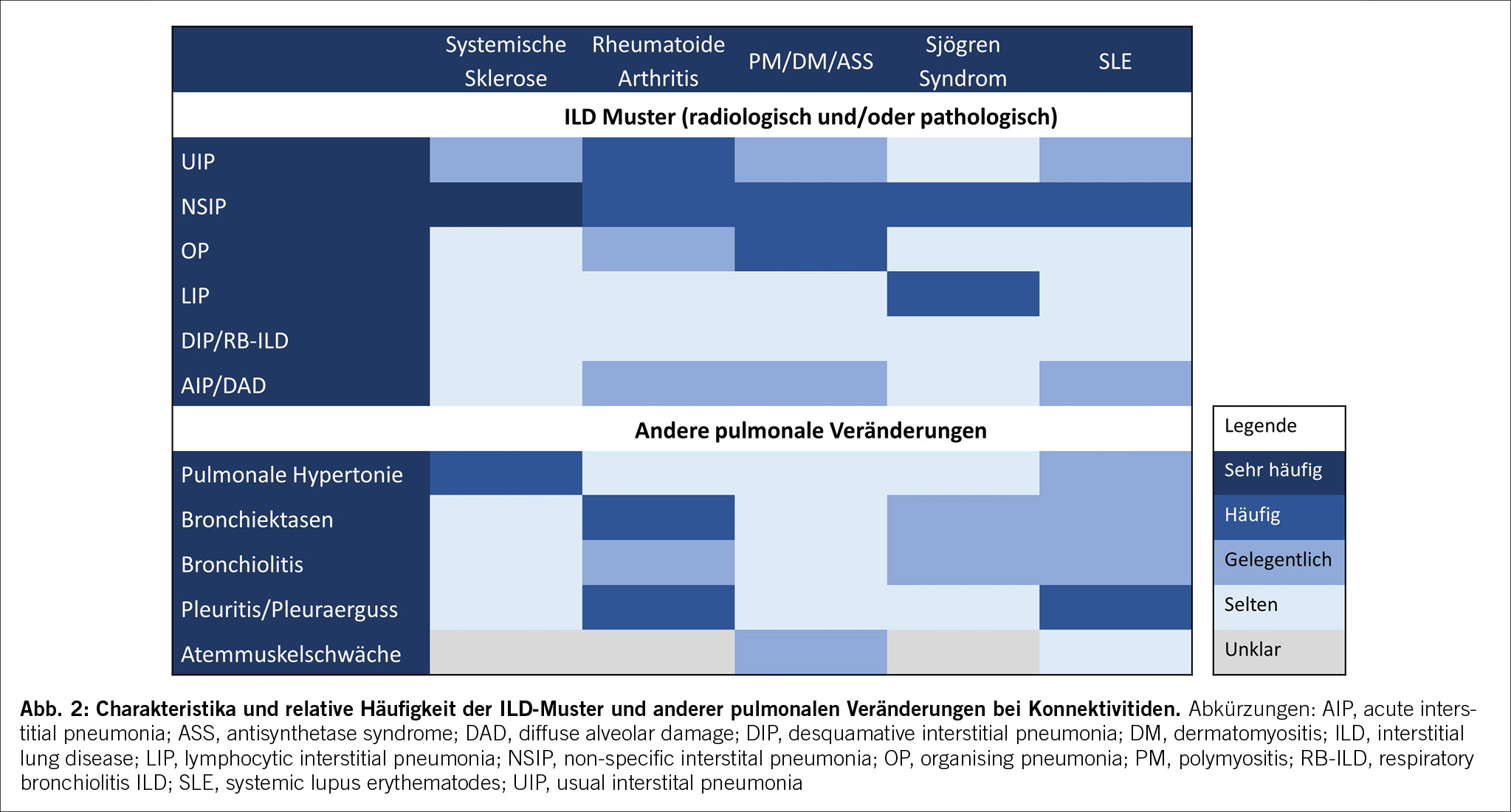
Die am häufigsten eingesetzten Therapien für die CTD-ILD sind systemische Glukokortikoide (GC), Mycophenolat Mofetil (MMF), Azathioprin (AZA), Methotrexat (MTX), Cyclophosamid (CYC), Rituximab (RTX) und Tocilizumab (TCZ).[63] Die Entscheidung ob und welche immunsuppressive Therapie indiziert ist, richtet sich nach der Grunderkrankung, der Aktivität der Erkrankung, der Art und dem Schweregrad des Organbefalls und der ILD Progression (Abb. 3).[4]
Der Einsatz von systemischen GC bei CTD-ILD sollte im Hinblick auf Wirkung und Nebenwirkungsprofil sorgfältig bedacht werden. Vorteil von GC sind die Verfügbarkeit und der schnelle und oft potente Therapieeffekt. Bei PM/DM-, RA- und Sjögren-Syndrom-ILD sind GC mit möglichst kurzer Therapiedauer und tiefer Dosierung empfohlen.[4],[64] Bei SSc sollte allerdings aufgrund des Risikos einer renalen Krise auf hochdosierte GC verzichtet werden. [65]
Die beste Datenlange zur immunsuppressiven Therapie findet sich zur SSc-ILD. Basierend auf der Evidenz aus den Scleroderma Lung Studies werden CYC und MMF häufig eingesetzt. Die Wirkung beider Substanzen ist ähnlich, die Toxizität aber insbesondere bei per oralem CYC hoch, sodass intravenöses CYC oder bezüglich Verträglichkeit oft MMF bevorzugt wird.[66-68] Eine intravenöse CYC Behandlung sollte insbesondere bei rasch progressiven CTD-ILD evaluiert werden. Retrospektive Studien zeigen, dass auch mit AZA bei Patient/-innen mit DM/PM-ILD eine Zunahme der FVC und DLCO mit weniger GC Bedarf erreicht werden kann. Unter AZA wurde häufiger eine Leukopenie und Erhöhung der Transaminasen beobachtet als unter MMF bei insgesamt 33% versus 14% der AZA und MMF Behandelten mit Nebenwirkungen.[69]. AZA ist kostengünstiger als MMF und auch eine gute Option bei Schwangeren. Um das Risiko einer Myelosuppression abzuschätzen,
kann vor Therapiebeginn die Aktivität der Thiopurinmethyltransferase bestimmt werden.[4] Als weiterer Antimetabolit kann auch MTX v.a. bei der RA eingesetzt werden und neuere Studiendaten zeigen, dass auch bei RA-ILD MTX eine sichere Therapieoption darstellt und sich positiv auf die Mortalität auswirken könnte.[62] [70]
Auch Biologika erhalten Einzug in die Therapie der CTD-ILD. Eine kleine randomisierte Placebo-kontrollierte Studie hatte in Patient/-innen mit SSc-ILD 24 Wochen nach Rituximab als sekundären Endpunkt eine im Vergleich zur Placebo –
Gruppe signifikant verbesserte FVC gezeigt.[71] Eine kürzlich publizierte Studie hatte den Effekt von RTX versus CYC in Patient/-innen mit progressiv fibrotischen CTD-ILDs untersucht und konnte in beiden Behandlungsgruppen zeigen, dass sich die FVC um ca. 100ml über 48 Wochen verbessert hatte, RTX war mit weniger Nebenwirkungen assoziiert als CYC.[72] Auch in retrospektiven Studien konnte unter Rituximab, bei SSc, MCTD, und beim Anti-Synthetase-Syndrom eine Stabilisierung der FVC,[73] und beim Sjögren-Syndrom eine radiologische und lungenfunktionelle (DLCO) Verbesserung gezeigt werden.[74] Basierend auf einer Phase III-Studie wurde Tocilizumab (TCZ) in den USA für die SSc-ILD zugelassen. Unter TCZ konnte eine signifikante Verlangsamung der Abnahme der FVC (sekundärer Endpunkt) in der SSc-ILD-Subpopulation nachgewiesen werden.[75] Eine Therapie mit TCZ kann vor allem bei SSc Patient/-innen mit persistierender Inflammation oder mit assoziierter Arthritis eingesetzt werden. Abatacept ist ein weiteres Biologikum mit vielversprechenden Resultaten aus Beobachtungsstudien,[76] wobei aktuell randomisiert-kontrollierte Studien für die RA-ILD und die Myositis assoziierte ILD laufen.
Die autologe Stammzelltransplantation stellt in ausgewählten Fällen eine gute Therapieoption dar. Bei der SSc zeigte eine randomisierte open-label Studie eine signifikante Verbesserung bezüglich Lunge und Haut. Zurückhaltung gegenüber der autologen Stammzelltransplantation bestand in der Vergangenheit wegen der hohen Toxizität-assoziierten Mortalität. Bei einer strengen Selektion der Population mit Ausschluss von Betroffenen (u.a.) mit FVC< 45% Soll, Pulmonaler Hypertonie oder linksventrikulärer Pumpfunktion < 40% lag die Mortalität bei 0% während und 2 Jahre nach autologer Stammzelltransplantation. [77-78]
Neben den immunsuppressiven Medikamenten stehen neu auch Antifibrotika zur Therapie der Konnektivitis-ILDs zur Verfügung. Nintedanib und Pirfenidon sind die zwei Antifibrotika welche seit ein paar Jahren für die Therapie der IPF zugelassen sind und Nintedanib ist seit 2020 auch für die Therapie der SSc-ILD zugelassen. Die SENSCIS hat als bislang grösste SSc-ILD Studie gezeigt, dass Nintedanib den Abfall der Vitalkapazität in etwa halbieren kann (jährlicher FVC-Verlust -52ml versus -93ml).[79] Auch wenn die absoluten Differenzen der FVC kleiner als bei der IPF sind, muss in Betracht gezogen werden, dass es sich häufig um junge Patient/-innen handelt bei welchen der kumulative FVC Verlust über die Jahre durchaus einen relevanten Verlust der Leistungsfähigkeit und Lebensqualität zur Folge haben kann. Die häufigste Nebenwirkung von Nintedanib ist eine bei SSc Patient/-innen ohnehin häufige (32%) Diarrhoe in 75% der Behandelten. Diesbezüglich ist eine sorgfältige Beratung bereits vor Therapiebeginn wichtig, damit Betroffene lernen, wie sie die Ernährung umstellen und falls nötig Loperamid einsetzen können. Die Studie hat auch gezeigt, dass der FVC-Verlauf unter der Kombination mit Mycophenolat mofetil am stabilsten und die Verträglichkeit in der Kombinationstherapie gleich ist.[80] Insbesondere bei progressiver CTD-ILD unter etablierter Immunsuppression sollte nicht nur bei der SSc-ILD, sondern auch bei der RA-ILD und der MCTD-ILD frühzeitig eine (zusätzliche) antifibrotische Therapie evaluiert werden.[81] Eine kürzlich publizierte Studie zu Pirfenidon bei RA-ILD hat zwar auf Grund von Rekrutierungsproblemen i.R. der COVID-19 Pandemie ihren primären Endpunkt verfehlt, hat aber gezeigt, dass RA-ILD Patienten unter Pirfenidon eine stabilere FVC als solche unter Placebo haben (-66ml versus -146ml). Die häufigste Nebenwirkung on Pirfenidon ist Nausea (53% versus 15% unter Placebo).[82]
Insgesamt ist die Entscheidung, wann welche Immunsuppression und wann eine antifibrotische Therapie eingesetzt werden soll, komplex und sollte in vielen Fällen durch ein multidisziplinäres Expertenteam getroffen werden. [13-14]
Nicht medikamentöse Therapien
Unabhängig von der Ursache der ILD ist auch bei der Konnektivitis-ILD ein konsequenter Rauchstopp, ausreichend Bewegung und eine ausgewogene Ernährung wichtig. Zudem sollten die Impfrichtlinien befolgt werden. Gerade junge Patient/-innen mit progressiver Konnektivitis-ILD sollten frühzeitig zur Abklärung einer Lungentransplantation zugewiesen werden, da dies in manchen Fällen der einzige kurative Ansatz bleibt. Ältere und multimorbide Patient/-innen hingegen profitieren oft von der Unterstützung eines Teams der Palliativmedizin, welches sich nicht nur um Aspekte des Lebensendes, sondern auch um eine optimale Symptomkontrolle kümmern kann.[45] Eine Heimsauerstofftherapie im Ruhezustand und bei körperlicher Belastung kann die Atemnot reduzieren und die körperliche Leistungsfähigkeit verbessern.[83] Zudem hat die pulmonale Rehabilitation ein eindrückliches Potential die Symptomatik, Lebensqualität und evtl. sogar die Prognose der ILD-Patient/-innen günstig zu beeinflussen[84],[85] Natürlich sollte bei Konnektivitis ein spezieller Fokus auf die muskuloskelettalen Einschränkungen gesetzt werden und auch die Rehabilitation interdisziplinär gestaltet werden. Da Komorbiditäten wie Pulmonale Hypertonie, Kardiopathien, Gastroösophagealer Reflux, Osteoporose und psychische Erkrankungen bei Konnektivitiden häufig sind, ist ein adäquates Management diesbezüglich wichtig. Auch eine häufige Polypharmazie ist gerade bei Polymorbiden Patient/-innen mit Frailty unbedingt zu beachten.[86]
Ausblick
Oft sind Diagnostik und Therapieentscheidung bei CTD-ILDs komplex und fordern eine multidisziplinäre Zusammenarbeit mit dem Ziel einer Verbesserung der Lebensqualität und des Langzeitüberlebens der Betroffenen.
Neue immunmodulatorische und antifibrotische Medikamente werden laufend in klinischen Studien untersucht und unser Armamentarium für die CTD-ILD Behandlung künftig erweitern. Neue diagnostische Vorgehensweisen und die Hilfe von künstlicher Intelligenz lassen auf eine präzisere Diagnostik und phänotypische Klassifikation hoffen[86], welche personalisierte Behandlungsstrategien für unsere Patient/-innen mit CTD-ILD ermöglichen können. Interdisziplinäre und interprofessionelle Abklärungs- und Betreuungsangebote werden an Zentren implementiert und ermöglichen eine effiziente Kommunikation und Koordination, sodass Betroffene von einer raschen, ganzheitlichen Abklärung, sowie Evidenz-basierte medikamentösen und nicht-medikamentösen Therapie profitieren können.
PD Dr. med. Sabina A. Guler, MHSc
Leitende Ärztin Universitätsklinik für
Pneumologie und Allergologie
Inselspital Bern
Freiburgstrasse, 3010 Bern
sabina.guler@insel.ch
Interessenskonflikt: Die Autoren haben keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
Universitätsklinik für
Pneumologie und Allergologie
Inselspital Bern
Freiburgstrasse, 3010 Bern
Die Autoren haben keine Interessenskonflikte im Zusammenhang mit diesem Artikel deklariert.
Literatur:
1. Duchemann B, Annesi-Maesano I, Jacobe de Naurois C, Sanyal S, Brillet PY, Brauner M, et al. Prevalence and incidence of interstitial lung diseases in a multi-ethnic county of Greater Paris. Eur Respir J. 2017 Aug;50(2):1602419.
2. Kaul B, Cottin V, Collard HR, Valenzuela C. Variability in Global Prevalence of Interstitial Lung Disease. Front Med. 2021;8:751181.
3. Fisher JH, Kolb M, Algamdi M, Morisset J, Johannson KA, Shapera S, et al. Baseline characteristics and comorbidities in the CAnadian REgistry for Pulmonary Fibrosis. BMC Pulm Med. 2019 Nov 27;19(1):223.
4. Jee AS, Sheehy R, Hopkins P, Corte TJ, Grainge C, Troy LK, et al. Diagnosis and management of connective tissue disease-associated interstitial lung disease in Australia and New Zealand: A position statement from the Thoracic Society of Australia and New Zealand. Respirol Carlton Vic. 2021 Jan;26(1):23–51.
5. Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972-2002. Ann Rheum Dis. 2007 Jul;66(7):940–4.
6. Hyldgaard C, Hilberg O, Pedersen AB, Ulrichsen SP, Løkke A, Bendstrup E, et al. A population-based cohort study of rheumatoid arthritis-associated interstitial lung disease: comorbidity and mortality. Ann Rheum Dis. 2017 Oct;76(10):1700–6.
7. Paulin F, Doyle TJ, Fletcher EA, Ascherman DP, Rosas IO. Rheumatoid Arthritis-Associated Interstitial Lung Disease and Idiopathic Pulmonary Fibrosis: Shared Mechanistic and Phenotypic Traits Suggest Overlapping Disease Mechanisms. Rev Investig Clin Organo Hosp Enfermedades Nutr. 2015;67(5):280–6.
8. Stanojevic S, Kaminsky DA, Miller MR, Thompson B, Aliverti A, Barjaktarevic I, et al. ERS/ATS technical standard on interpretive strategies for routine lung function tests. Eur Respir J. 2022 Jul;60(1):2101499.
9. Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2022 May 1;205(9):e18–47.
10. Travis WD, Costabel U, Hansell DM, King TE, Lynch DA, Nicholson AG, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013 Sep 15;188(6):733–48.
11. Adams SJ, Stone E, Baldwin DR, Vliegenthart R, Lee P, Fintelmann FJ. Lung cancer screening. Lancet Lond Engl. 2023 Feb 4;401(10374):390–408.
12. Chatzidionysiou K, di Giuseppe D, Soderling J, Catrina A, Askling J. Risk of lung cancer in rheumatoid arthritis and in relation to autoantibody positivity and smoking. RMD Open. 2022 Oct;8(2):e002465.
13. Walsh SLF, Wells AU, Desai SR, Poletti V, Piciucchi S, Dubini A, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir Med. 2016 Jul;4(7):557–65.
14. Guler SA, Berezowska SA, Christe A, Geiser T, Funke-Chambour M. Multidisciplinary discussion for diagnosis of interstitial lung disease in real life. Swiss Med Wkly. 2016;146:w14318.
15. Meyer KC, Raghu G, Baughman RP, Brown KK, Costabel U, du Bois RM, et al. An official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med. 2012 May 1;185(9):1004–14.
16. Goulabchand R, Malafaye N, Jacot W, Witkowski Durand Viel P, Morel J, Lukas C, et al. Cancer incidence in primary Sjögren’s syndrome: Data from the French hospitalization database. Autoimmun Rev. 2021 Dec;20(12):102987.
17. Lis-Święty A, Widuchowska M, Brzezińska-Wcisło L, Kucharz E. High acute phase protein levels correlate with pulmonary and skin involvement in patients with diffuse systemic sclerosis. J Int Med Res. 2018 Apr;46(4):1634–9.
18. Mitev A, Christ L, Feldmann D, Binder M, Möller K, Kanne AM, et al. Inflammatory stays inflammatory: a subgroup of systemic sclerosis characterized by high morbidity and inflammatory resistance to cyclophosphamide. Arthritis Res Ther. 2019 Dec 2;21(1):262.
19. Jacobi C, Wildemann B. Labordiagnostik von Kollagenosen unter besonderer Berücksichtigung neurologischer Manifestationen / Labaratory diagnosis of connective tissue diseases with a focus on neurological manifestations. LaboratoriumsMedizin. 2006 Jan 1;30(5):280–8.
20. Kondoh Y, Makino S, Ogura T, Suda T, Tomioka H, Amano H, et al. 2020 guide for the diagnosis and treatment of interstitial lung disease associated with connective tissue disease. Respir Investig. 2021 Nov;59(6):709–40.
21. Forslind E. [Employment agencies help the USA recruit Swedish nurses]. Vardfacket. 1992 Aug 27;16(13):7.
22. Doyle TJ, Patel AS, Hatabu H, Nishino M, Wu G, Osorio JC, et al. Detection of Rheumatoid Arthritis-Interstitial Lung Disease Is Enhanced by Serum Biomarkers. Am J Respir Crit Care Med. 2015 Jun 15;191(12):1403–12.
23. Lingg G, Schorn C. [Differential diagnosis of rheumatic diseases]. Radiol. 2006 May;46(5):354–64.
24. Hauer RW, Schmidt WA, Bohl-Bühler M, Banzer D, Mellerowicz H, Sattler H, et al. [Technique and value of arthrosonography in rheumatologic diagnosis. 1: Ultrasound diagnosis of the knee joint]. Z Rheumatol. 2001 Jun;60(3):139–47.
25. Courtney P, Doherty M. Joint aspiration and injection and synovial fluid analysis. Best Pract Res Clin Rheumatol. 2013 Apr;27(2):137–69.
26. van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an American college of rheumatology/European league against rheumatism collaborative initiative. Ann Rheum Dis. 2013 Nov;72(11):1747–55.
27. J.S. Parker M, W. McGill N. The Established and Evolving Role of Nailfold Capillaroscopy in Connective-Tissue Disease. In: Takeda A, editor. Connective Tissue Disease – Current State of the Art [Internet]. IntechOpen; 2020 [cited 2023 Feb 12]. Available from: https://www.intechopen.com/books/connective-tissue-disease-current-state-of-the-art/the-established-and-evolving-role-of-nailfold-capillaroscopy-in-connective-tissue-disease
28. Castellví I, Simeón-Aznar CP, Sarmiento M, Fortuna A, Mayos M, Geli C, et al. Association between nailfold capillaroscopy findings and pulmonary function tests in patients with systemic sclerosis. J Rheumatol. 2015 Feb;42(2):222–7.
29. Stefanski AL, Tomiak C, Pleyer U, Dietrich T, Burmester GR, Dörner T. The Diagnosis and Treatment of Sjögren’s Syndrome. Dtsch Arzteblatt Int. 2017 May 26;114(20):354–61.
30. Hoffmann-Vold AM, Fretheim H, Halse AK, Seip M, Bitter H, Wallenius M, et al. Tracking Impact of Interstitial Lung Disease in Systemic Sclerosis in a Complete Nationwide Cohort. Am J Respir Crit Care Med. 2019 Nov 15;200(10):1258–66.
31. Khanna D, Tashkin DP, Denton CP, Renzoni EA, Desai SR, Varga J. Etiology, Risk Factors, and Biomarkers in Systemic Sclerosis with Interstitial Lung Disease. Am J Respir Crit Care Med. 2020 Mar 15;201(6):650–60.
32. Hoffmann-Vold AM, Allanore Y, Alves M, Brunborg C, Airó P, Ananieva LP, et al. Progressive interstitial lung disease in patients with systemic sclerosis-associated interstitial lung disease in the EUSTAR database. Ann Rheum Dis. 2021 Feb;80(2):219–27.
33. Guler SA, Winstone TA, Murphy D, Hague C, Soon J, Sulaiman N, et al. Does Systemic Sclerosis-associated Interstitial Lung Disease Burn Out? Specific Phenotypes of Disease Progression. Ann Am Thorac Soc. 2018 Dec;15(12):1427–33.
34. Nihtyanova SI, Schreiber BE, Ong VH, Rosenberg D, Moinzadeh P, Coghlan JG, et al. Prediction of pulmonary complications and long-term survival in systemic sclerosis. Arthritis Rheumatol Hoboken NJ. 2014 Jun;66(6):1625–35.
35. Distler O, Assassi S, Cottin V, Cutolo M, Danoff SK, Denton CP, et al. Predictors of progression in systemic sclerosis patients with interstitial lung disease. Eur Respir J. 2020 May;55(5):1902026.
36. Roman S, Hot A, Fabien N, Cordier JF, Miossec P, Ninet J, et al. Esophageal dysmotility associated with systemic sclerosis: a high-resolution manometry study. Dis Esophagus Off J Int Soc Dis Esophagus. 2011 Jul;24(5):299–304.
37. Hershcovici T, Jha LK, Johnson T, Gerson L, Stave C, Malo J, et al. Systematic review: the relationship between interstitial lung diseases and gastro-oesophageal reflux disease. Aliment Pharmacol Ther. 2011 Dec;34(11–12):1295–305.
38. Tucker AE, Perin J, Volkmann ER, Abdi T, Shah AA, Pandolfino J, et al. Associations between Patterns of Esophageal Dysmotility and Extra-Intestinal Features in Patients with Systemic Sclerosis. Arthritis Care Res. 2022 Dec 28;
39. Jeganathan N, Nguyen E, Sathananthan M. Rheumatoid Arthritis and Associated Interstitial Lung Disease: Mortality Rates and Trends. Ann Am Thorac Soc. 2021 Dec;18(12):1970–7.
40. Hunter TM, Boytsov NN, Zhang X, Schroeder K, Michaud K, Araujo AB. Prevalence of rheumatoid arthritis in the United States adult population in healthcare claims databases, 2004-2014. Rheumatol Int. 2017 Sep;37(9):1551–7.
41. Hyldgaard C, Hilberg O, Pedersen AB, Ulrichsen SP, Løkke A, Bendstrup E, et al. A population-based cohort study of rheumatoid arthritis-associated interstitial lung disease: comorbidity and mortality. Ann Rheum Dis. 2017 Oct;76(10):1700–6.
42. Reid P, Guler SA. Mortality Trends in Rheumatoid Arthritis: Zooming in on Interstitial Lung Disease. Ann Am Thorac Soc. 2021 Dec;18(12):1953–4.
43. Solomon JJ, Chung JH, Cosgrove GP, Demoruelle MK, Fernandez-Perez ER, Fischer A, et al. Predictors of mortality in rheumatoid arthritis-associated interstitial lung disease. Eur Respir J. 2016 Feb;47(2):588–96.
44. Kadura S, Raghu G. Rheumatoid arthritis-interstitial lung disease: manifestations and current concepts in pathogenesis and management. Eur Respir Rev Off J Eur Respir Soc. 2021 Jun 30;30(160):210011.
45. Raghu G, Remy-Jardin M, Richeldi L, Thomson CC, Inoue Y, Johkoh T, et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2022 May 1;205(9):e18–47.
46. Sun KY, Fan Y, Wang YX, Zhong YJ, Wang GF. Prevalence of interstitial lung disease in polymyositis and dermatomyositis: A meta-analysis from 2000 to 2020. Semin Arthritis Rheum. 2021 Feb;51(1):175–91.
47. Braun NM, Arora NS, Rochester DF. Respiratory muscle and pulmonary function in polymyositis and other proximal myopathies. Thorax. 1983 Aug;38(8):616–23.
48. Luppi F, Sebastiani M, Silva M, Sverzellati N, Cavazza A, Salvarani C, et al. Interstitial lung disease in Sjögren’s syndrome: a clinical review. Clin Exp Rheumatol. 2020;38 Suppl 126(4):291–300.
49. Kelly C, Gardiner P, Pal B, Griffiths I. Lung function in primary Sjögren’s syndrome: a cross sectional and longitudinal study. Thorax. 1991 Mar;46(3):180–3.
50. Chen MH, Chou HP, Lai CC, Chen YD, Chen MH, Lin HY, et al. Lung involvement in primary Sjögren’s syndrome: Correlation between high-resolution computed tomography score and mortality. J Chin Med Assoc JCMA. 2014 Feb;77(2):75–82.
51. Enomoto Y, Takemura T, Hagiwara E, Iwasawa T, Fukuda Y, Yanagawa N, et al. Prognostic factors in interstitial lung disease associated with primary Sjögren’s syndrome: a retrospective analysis of 33 pathologically-proven cases. PloS One. 2013;8(9):e73774.
52. Flament T, Bigot A, Chaigne B, Henique H, Diot E, Marchand-Adam S. Pulmonary manifestations of Sjögren’s syndrome. Eur Respir Rev Off J Eur Respir Soc. 2016 Jun;25(140):110–23.
53. Gunnarsson R, Aaløkken TM, Molberg Ø, Lund MB, Mynarek GK, Lexberg AS, et al. Prevalence and severity of interstitial lung disease in mixed connective tissue disease: a nationwide, cross-sectional study. Ann Rheum Dis. 2012 Dec;71(12):1966–72.
54. Gunnarsson R, Hetlevik SO, Lilleby V, Molberg Ø. Mixed connective tissue disease. Best Pract Res Clin Rheumatol. 2016 Feb;30(1):95–111.
55. Ryerson CJ, Corte TJ, Myers JL, Walsh SLF, Guler SA. A contemporary practical approach to the multidisciplinary management of unclassifiable interstitial lung disease. Eur Respir J. 2021 Dec;58(6):2100276.
56. Guler SA, Ellison K, Algamdi M, Collard HR, Ryerson CJ. Heterogeneity in Unclassifiable Interstitial Lung Disease. A Systematic Review and Meta-Analysis. Ann Am Thorac Soc. 2018 Jul;15(7):854–63.
57. Glenn LM, Pugashetti JV, Oldham J, Corte TJ. Interstitial pneumonia with autoimmune features: from research classification to diagnosis. Curr Opin Pulm Med. 2021 Sep 1;27(5):374–87.
58. Oldham JM, Adegunsoye A, Valenzi E, Lee C, Witt L, Chen L, et al. Characterisation of patients with interstitial pneumonia with autoimmune features. Eur Respir J. 2016 Jun;47(6):1767–75.
59. Wong AW, Ryerson CJ, Guler SA. Progression of fibrosing interstitial lung disease. Respir Res. 2020 Jan 29;21(1):32.
60. Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am J Respir Crit Care Med. 2018 Sep 1;198(5):e44–68.
61. Kremer JM, Alarcón GS, Weinblatt ME, Kaymakcian MV, Macaluso M, Cannon GW, et al. Clinical, laboratory, radiographic, and histopathologic features of methotrexate-associated lung injury in patients with rheumatoid arthritis: a multicenter study with literature review. Arthritis Rheum. 1997 Oct;40(10):1829–37.
62. Kiely P, Busby AD, Nikiphorou E, Sullivan K, Walsh DA, Creamer P, et al. Is incident rheumatoid arthritis interstitial lung disease associated with methotrexate treatment? Results from a multivariate analysis in the ERAS and ERAN inception cohorts. BMJ Open. 2019 May 5;9(5):e028466.
63. Maher TM. Immunosuppression for connective tissue disease-related pulmonary disease. Semin Respir Crit Care Med. 2014 Apr;35(2):265–73.
64. Ramos-Casals M, Brito-Zerón P, Bombardieri S, Bootsma H, De Vita S, Dörner T, et al. EULAR recommendations for the management of Sjögren’s syndrome with topical and systemic therapies. Ann Rheum Dis. 2020 Jan;79(1):3–18.
65. Hoffmann-Vold AM, Maher TM, Philpot EE, Ashrafzadeh A, Barake R, Barsotti S, et al. The identification and management of interstitial lung disease in systemic sclerosis: evidence-based European consensus statements. Lancet Rheumatol. 2020 Feb;2(2):e71–83.
66. Tashkin DP, Roth MD, Clements PJ, Furst DE, Khanna D, Kleerup EC, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med. 2016 Sep;4(9):708–19.
67. Tashkin DP, Elashoff R, Clements PJ, Roth MD, Furst DE, Silver RM, et al. Effects of 1-year treatment with cyclophosphamide on outcomes at 2 years in scleroderma lung disease. Am J Respir Crit Care Med. 2007 Nov 15;176(10):1026–34.
68. Barnes H, Holland AE, Westall GP, Goh NS, Glaspole IN. Cyclophosphamide for connective tissue disease-associated interstitial lung disease. Cochrane Database Syst Rev. 2018 Jan 3;1(1):CD010908.
69. Huapaya JA, Silhan L, Pinal-Fernandez I, Casal-Dominguez M, Johnson C, Albayda J, et al. Long-Term Treatment With Azathioprine and Mycophenolate Mofetil for Myositis-Related Interstitial Lung Disease. Chest. 2019 Nov;156(5):896–906.
70. Rojas-Serrano J, Herrera-Bringas D, Pérez-Román DI, Pérez-Dorame R, Mateos-Toledo H, Mejía M. Rheumatoid arthritis-related interstitial lung disease (RA-ILD): methotrexate and the severity of lung disease are associated to prognosis. Clin Rheumatol. 2017 Jul;36(7):1493–500.
71. Ebata S, Yoshizaki A, Oba K, Kashiwabara K, Ueda K, Uemura Y, et al. Safety and efficacy of rituximab in systemic sclerosis (DESIRES): a double-blind, investigator-initiated, randomised, placebo-controlled trial. Lancet Rheumatol. 2021 Jul;3(7):e489–97.
72. Maher TM, Tudor VA, Saunders P, Gibbons MA, Fletcher SV, Denton CP, et al. Rituximab versus intravenous cyclophosphamide in patients with connective tissue disease-associated interstitial lung disease in the UK (RECITAL): a double-blind, double-dummy, randomised, controlled, phase 2b trial. Lancet Respir Med. 2023 Jan;11(1):45–54.
73. Lepri G, Avouac J, Airò P, Anguita Santos F, Bellando-Randone S, Blagojevic J, et al. Effects of rituximab in connective tissue disorders related interstitial lung disease. Clin Exp Rheumatol. 2016;34 Suppl 100(5):181–5.
74. Chen MH, Chen CK, Chou HP, Chen MH, Tsai CY, Chang DM. Rituximab therapy in primary Sjögren’s syndrome with interstitial lung disease: a retrospective cohort study. Clin Exp Rheumatol. 2016;34(6):1077–84.
75. Khanna D, Lin CJF, Furst DE, Goldin J, Kim G, Kuwana M, et al. Tocilizumab in systemic sclerosis: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir Med. 2020 Oct;8(10):963–74.
76. Vicente-Rabaneda EF, Atienza-Mateo B, Blanco R, Cavagna L, Ancochea J, Castañeda S, et al. Efficacy and safety of abatacept in interstitial lung disease of rheumatoid arthritis: A systematic literature review. Autoimmun Rev. 2021 Jun;20(6):102830.
77. Burt RK, Shah SJ, Dill K, Grant T, Gheorghiade M, Schroeder J, et al. Autologous non-myeloablative haemopoietic stem-cell transplantation compared with pulse cyclophosphamide once per month for systemic sclerosis (ASSIST): an open-label, randomised phase 2 trial. Lancet Lond Engl. 2011 Aug 6;378(9790):498–506.
78. van Laar JM, Farge D, Sont JK, Naraghi K, Marjanovic Z, Larghero J, et al. Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial. JAMA. 2014 Jun 25;311(24):2490–8.
79. Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, Mayes MD, et al. Nintedanib for Systemic Sclerosis-Associated Interstitial Lung Disease. N Engl J Med. 2019 Jun 27;380(26):2518–28.
80. Jehle J, Bornikoel K, Neuhaus KL, Spiller P, Sauer G. [Left ventricular elasticity and stress pressure in the small circulation before and after coronary bypass]. Verh Dtsch Ges Inn Med. 1978;(84):670–3.
81. Wells AU, Flaherty KR, Brown KK, Inoue Y, Devaraj A, Richeldi L, et al. Nintedanib in patients with progressive fibrosing interstitial lung diseases-subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: a randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir Med. 2020 May;8(5):453–60.
82. Solomon JJ, Danoff SK, Woodhead FA, Hurwitz S, Maurer R, Glaspole I, et al. Safety, tolerability, and efficacy of pirfenidone in patients with rheumatoid arthritis-associated interstitial lung disease: a randomised, double-blind, placebo-controlled, phase 2 study. Lancet Respir Med. 2023 Jan;11(1):87–96.
83. Bell EC, Cox NS, Goh N, Glaspole I, Westall GP, Watson A, et al. Oxygen therapy for interstitial lung disease: a systematic review. Eur Respir Rev Off J Eur Respir Soc. 2017 Jan;26(143):160080.
84. Dowman LM, McDonald CF, Hill CJ, Lee AL, Barker K, Boote C, et al. The evidence of benefits of exercise training in interstitial lung disease: a randomised controlled trial. Thorax. 2017 Jul;72(7):610–9.
85. Guler SA, Hur SA, Stickland MK, Brun P, Bovet L, Holland AE, et al. Survival after inpatient or outpatient pulmonary rehabilitation in patients with fibrotic interstitial lung disease: a multicentre retrospective cohort study. Thorax. 2022 Jun;77(6):589–95.
86. Guler SA, Kwan JM, Winstone TA, Milne KM, Dunne JV, Wilcox PG, et al. Severity and features of frailty in systemic sclerosis-associated interstitial lung disease. Respir Med. 2017 Aug;129:1–7.
87. Wells AU, Denton CP. Interstitial lung disease in connective tissue disease–mechanisms and management. Nat Rev Rheumatol. 2014 Dec;10(12):728–39.
88. www.pneumotox.com
Therapeutische Umschau
- Vol. 81
- Ausgabe 1
- Februar 2024