Zusammenfassung: Die 2023 veröffentlichten Behandlungsrichtlinien der Europäischen Gesellschaft für Kardiologie (ESC) für das Management der Kardiomyopathien behandeln erstmals alle Kardiomyopathien in einem Dokument. Im Fokus stehen ein Phänotyp-orientierter diagnostischer Zugang, multimodale Bildgebung und genetische Tests, um eine möglichst genaue Diagnose zu stellen. Darüber hinaus werden neue Empfehlungen zur Risikostratifizierung des plötzlichen Herztods bei verschiedenen Kardiomyopathie-Phänotypen gegeben. Hierbei hat die MRI- und Genetik-Diagnostik erheblich an Bedeutung gewonnen. Empfehlungen für das umfassende klinische und genetische Kaskadenscreening bei Verwandten von Personen mit Kardiomyopathien wurden überarbeitet. Im vorliegenden Artikel werden die wichtigsten Neuerungen nach einem praxisorientierten Ansatz vorgestellt.
Cardiomyopathies: a practical approach to the assessment and management of patients and their families
Abstract: The new 2023 European Society of Cardiology (ESC) Guidelines for the management of cardiomyopathies addresses all cardiomyopathies in a single document for the first time. The focus is on a phenotype-oriented diagnostic approach, multimodal imaging and genetic testing to establish the most accurate diagnosis possible. Additionally, new recommendations for risk stratification for sudden cardiac death in various cardiomyopathy phenotypes are provided. MRI and genetic testing have significantly gained importance in this context. Recommendations for comprehensive clinical and genetic cascade screening in relatives of individuals with cardiomyopathies have been revised. This article presents the most important innovations of these guidelines in a practice-oriented approach.
Einleitung
Kardiomyopathien (KMP) stellen eine vielseitige Gruppe von Herzmuskelerkrankungen dar, die oft mit Herzinsuffizienzsymptomen verbunden sind und mit einem erhöhten Risiko für Rhythmusstörungen inklusive plötzlichem Herztod (SCD) einhergehen. Die meisten dieser Erkrankungen kommen familiär gehäuft vor, was nicht nur für die betroffenen Patienten, sondern auch für deren Familien Implikationen hat. Die häufigsten und bekanntesten primären KMP umfassen die hypertrophe Kardiomyopathie (HCM), die dilatative Kardiomyopathie (DCM) und die rechtsventrikuläre arrhythmogene Kardiomyopathie (ARVC) (1). Durch die Fortschritte in der kardialen Bildgebung, der Genetik und vermehrter Sensibilisierung der Ärzte werden diese Erkrankungen immer häufiger erkannt, und die betroffenen Patienten und Familien bedürfen einer entsprechenden interdisziplinären Betreuung.
Wie in der Medizin im Allgemeinen ist der Bereich der KMP von der Erstbeschreibung der jeweiligen Formen beeinflusst durch Forschungsergebnisse und technische Erneuerungen historisch gewachsen. Am besten lässt sich das anhand der HCM aufweisen, welche schon früh intensiv erforscht wurde. Die HCM wurde in der medizinischen Fachwelt erstmals 1958 wahrgenommen, als ein Londoner Pathologe – Dr. Donald Teare – die Erkrankung in einer Familie mit vielen plötzlichen Todesfällen im jungen Alter als «Asymmetrical Hypertrophy of the Heart» beschrieben hatte (2). Kurz darauf – nur 3 Jahre später – hat Prof. Morrow erstmals eine chirurgische Myektomie zur Behandlung der Obstruktion des linksventrikulären Ausflusstrakts (LVOT) durchgeführt (3). Weitere Meilensteine in der Behandlung und Diagnostik der hypertrophen Kardiomyopathie stellen die Entwicklung der Echokardiographie (1972), das Herz-MRI (2000), des intrakardialen Cardioverter-Defibrillators (ICD) (1980), Septalalkoholablation (1984) und der Genetik in den 1990-ern dar. Diese Meilensteine beeinflussen das Krankheitsverständnis, die diagnostischen und therapeutischen Möglichkeiten nachhaltig und sind in einem stetigen Wandel begriffen.
Wichtig ist zu erwähnen, dass KMP als Herzmuskelerkrankungen von koronaren, hypertensiven, valvulären und kongenitalen Herzerkrankungen differenziert werden. Da in der Allgemeinbevölkerung sowohl die arterielle Hypertonie als auch die koronare Herzkrankheit sehr weit verbreitet sind, können diese mit den primären KMP aber auch koexistieren. Im Sommer 2023 hat die Europäische Gesellschaft für Kardiologie (ESC) neue Empfehlungen zur Abklärung und zum Management von KMP veröffentlicht, was in dieser Ausgabe zum Anlass genommen wurde, eine pragmatische Herangehensweise für die Betroffenen zu erarbeiten (4).
Einteilung der KMP gemäss ESC-Behandlungsrichtlinien 2023
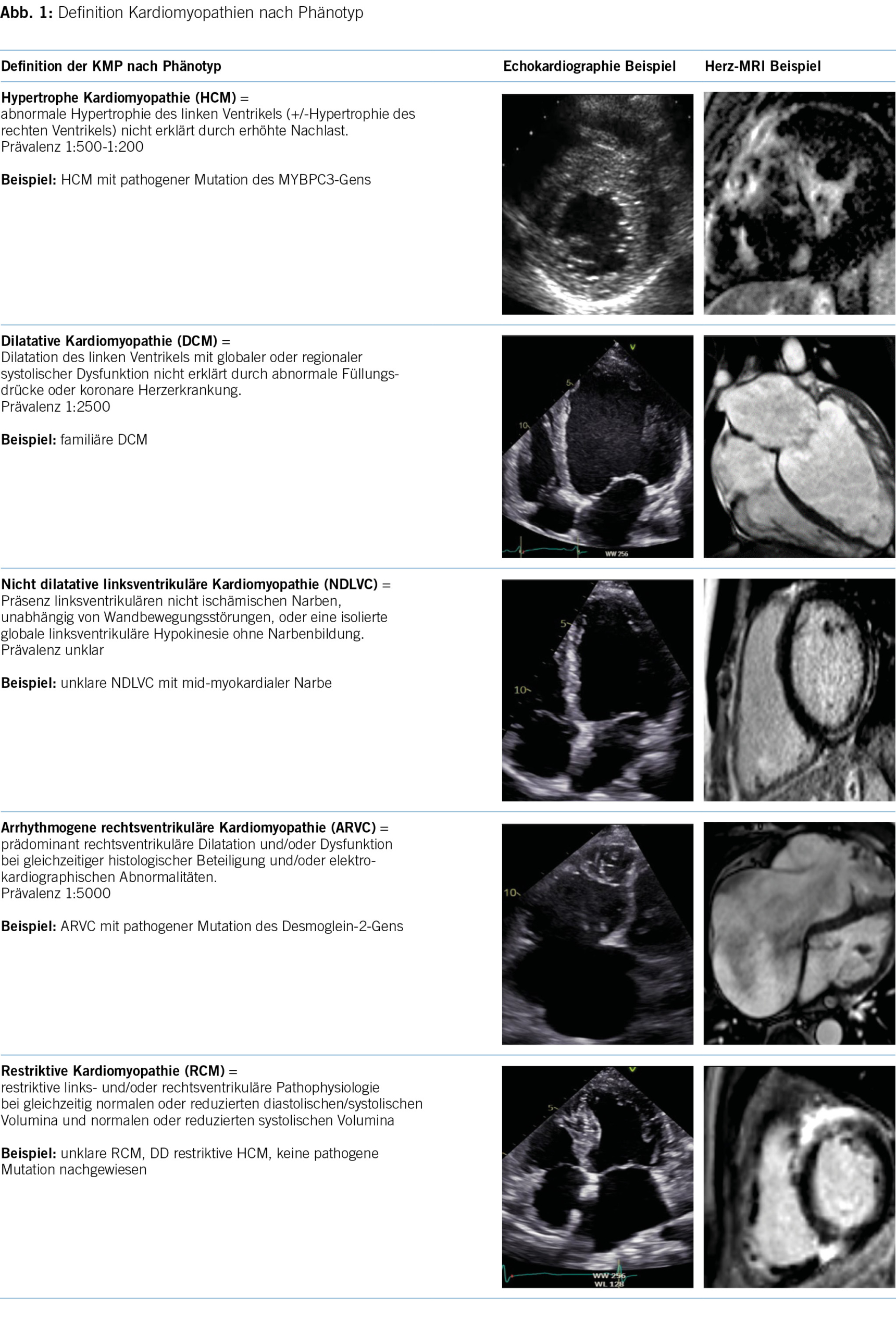
Die Kernaussage der 2023 KMP-Leitlinien besteht vor allem darin, dass bei den behandelnden Ärzten ein sogenanntes cardiomyopathy mindset entwickelt wird und die Ärzte sich nicht nur auf den morphologischen Phänotyp konzentrieren, sondern am Anfang der Patient mit seiner klinischen Präsentation steht (5). Im Rahmen der initialen Abklärung sollen dann auch die differentialdiagnostischen Überlegungen im Zentrum stehen, denn die korrekte Diagnose hat mittlerweile immer mehr Implikationen für die Prognose und therapeutischen Optionen der Patienten und deren Familien. Die ESC 2023 KMP-Leitlinie integriert die bestehenden vier Phänotypen und erweitert sie um einen neuen, fünften Phänotyp namens «nichtdilatierte linksventrikuläre Kardiomyopathie» (NDLVC). Diese Kategorie umfasst Patienten mit isolierter linksventrikulärer Dysfunktion ohne Narbenbildung sowie Patienten mit nicht ischämischer Narbenbildung, unabhängig von einer systolischen Dysfunktion. Dadurch wird ermöglicht, Phänotypen zu berücksichtigen, die trotz Vorliegen einer Myokarderkrankung nicht den Definitionen der anderen Klassen entsprechen (Abbildung 1). Eine wichtige Änderung bei der Einteilung der KMP ist die Beibehaltung des Phänotyps «ARVC», um die ursprüngliche Definition zu beschreiben, bei der die Dilatation des Ventrikels und/oder Wandbewegungsstörungen hauptsächlich auf den rechten Ventrikel beschränkt sind, mit oder ohne Beteiligung des linken Ventrikels (6). Die Verwendung des Sammelbegriffs «arrhythmogene KMP (ACM)» wird nicht anerkannt, und bei einer überwiegenden linksventrikulären Erkrankung sollen die Empfehlungen für NDLVC angewendet werden.
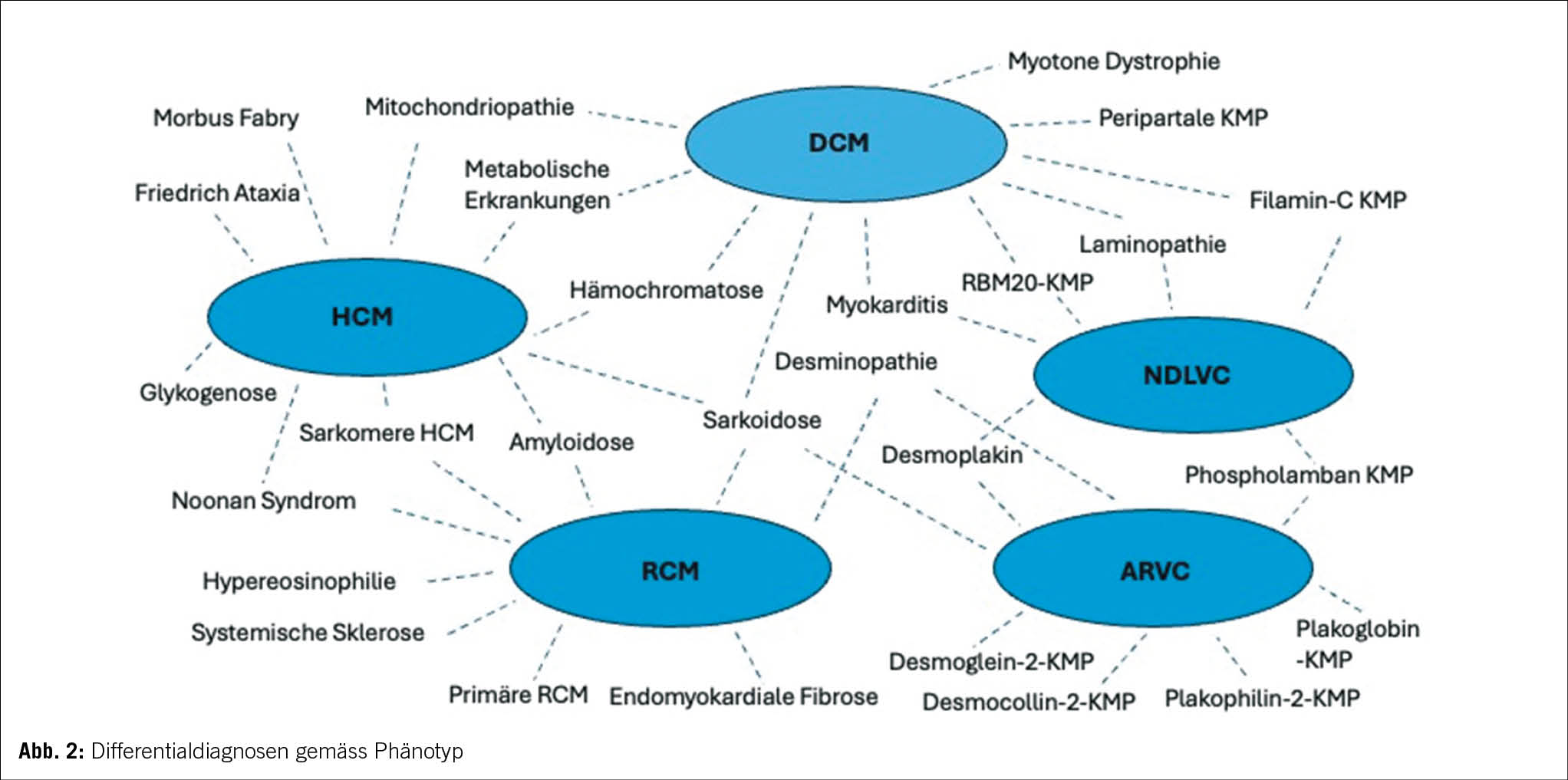
Eine weitere Änderung ist die Abschaffung des Begriffes «non-compaction KMP» als eigenständige KMP. Angesichts des Mangels an morphometrischen Beweisen für ventrikuläre Verdichtung beim Menschen wird der Begriff «Hypertrabekulierung» anstelle von LVNC empfohlen, insbesondere wenn das Phänomen reversibel ist oder klar im Erwachsenenalter auftritt (7, 8). Hinzu kommt, dass sowohl die phänotypische als auch genetische Überlappung mit der DCM und HCM substanziell ist (9). In diesem Zusammenhang bleibt zu erwähnen, dass auch innerhalb derselben Familie mit sehr ähnlichem genetischen Hintergrund verschiedene KMP-Phänotypen auftreten können (10). Dies wird aktuell weiter untersucht, und man vermutet epigenetische wie auch Umweltfaktoren, die jeweils für die Ausbildung der jeweiligen Phänotypen verantwortlich sind. Ebenfalls kann der Krankheitsverlauf bei einem einzelnen Patienten eine Entwicklung von einem KMP-Phänotyp zu einem anderen einschliessen. Die Arbeitsgruppe schlägt einen Ansatz für die Krankheitsnomenklatur und Diagnose vor, der auf dem vorherrschenden kardialen Phänotyp bei der Präsentation basiert. Dennoch ist der Genotyp wichtig für die diagnostische Abklärung, Therapieentscheidungen und Nachsorge. Obwohl die genetischen KMP der Schwerpunkt der neuen Leitlinien sind, wird weiterhin ein systematischer Ansatz ausgehend vom morpho-funktionellem Phänotyp bis zu Erreichen einer präzisen Diagnostik vorgeschlagen. Dabei werden auch nicht genetische KMP, wie zum Beispiel entzündlich, toxisch und multisystemisch, berücksichtigt. Abbildung 2 bietet einen Überblick über mögliche Differentialdiagnosen einschliesslich Phänokopien gemäss morpho-funktionellem Phänotyp. Wichtig ist zu vermerken, dass die endgültige Diagnose idealerweise die Ätiologie neben dem Phänotyp beschreiben sollte. In der Betreuung dieser Patienten und Familien wird es weiterhin so sein, dass für einen substanziellen Anteil der Betroffenen keine eindeutige Diagnose vorliegen wird, die man weiterhin pragmatisch nach dem Ansatz des vorliegenden Phänotyps behandeln wird.

Diagnostik
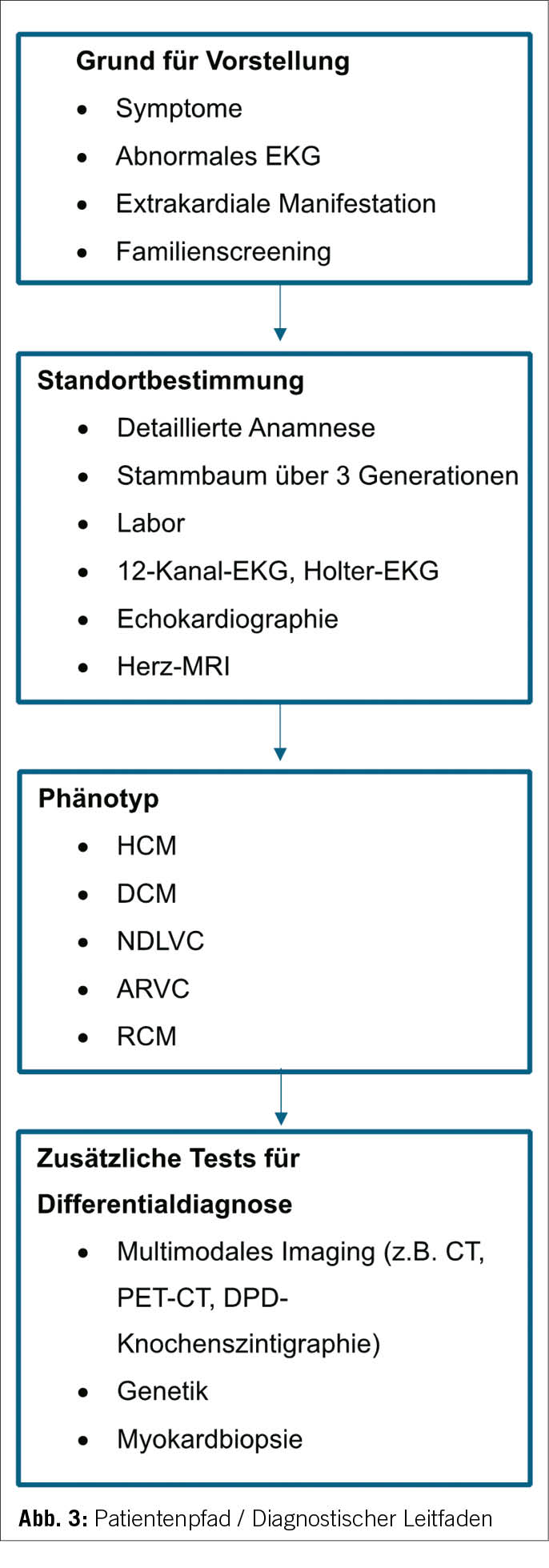
Patientenpfad für eine Phänotyp-orientierte diagnostische Überlegung
Gleichzeitig wurde ein konzeptueller Rahmen für Diagnose und Behandlung bereitgestellt. Das Konzept des Patientenwegs mit einem umfassenden Ansatz in der Patientenversorgung beginnt mit der ersten klinischen Vorstellung und durchläuft verschiedene klinische Untersuchungen inklusive kardiale Bildgebung, um den offensichtlichsten Phänotyp der KMP zu identifizieren und darauf basierend differentialdiagnostische Überlegungen und weiterführende Abklärungen in die Wege zu leiten (Abbildung 3).
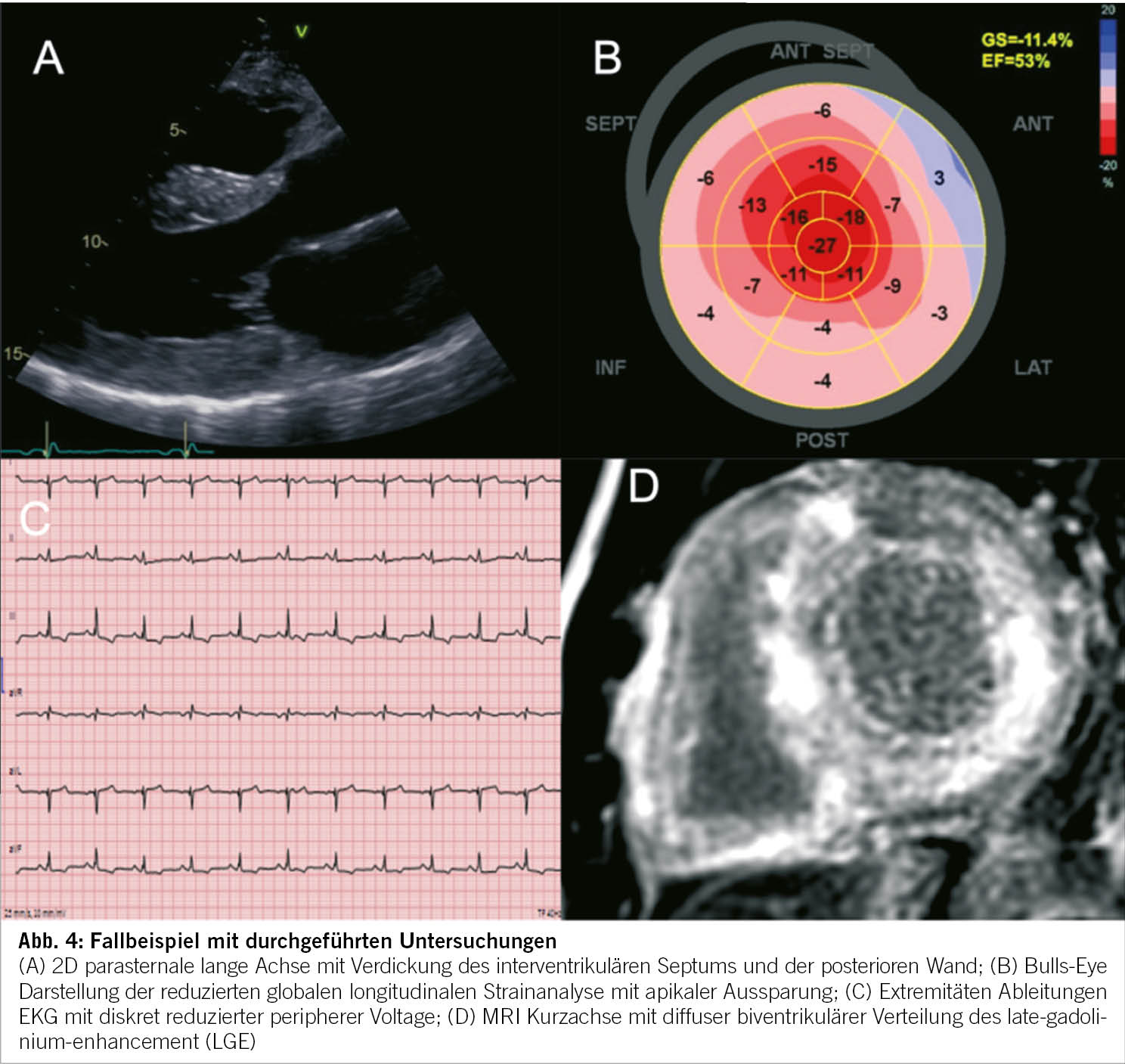
Folgendes Fallbeispiel eines 49-jährigen, männlichen Patienten mit progredienter Dyspnoe ist eine Illustration des diagnostischen Leitfadens. In der medizinischen Vorgeschichte sind eine unklare Niereninsuffizienz und ein bilaterales Karpaltunnelsyndrom bekannt. Die Familienanamnese ist bezüglich kardiovaskulärer Erkrankungen unauffällig. In der körperlichen Untersuchung fallen vor allem ein verbreiteter Herzspitzenstoss und ein vierter Herzton auf. Die pathologischen Werte in der Laboruntersuchung sind: Kreatinin 130umol/l, Troponin 46ng/l, NTproBNP 1700ng/l. Im 12-Kanal-EKG werden ein normokarder Sinusrhythmus, normale Zeitindices, ein Linkslagetyp, ST-Senkungen inferior und diskrete periphere Niedervoltage beobachtet. Die Echokardiographie zeigte einen konzentrisch verdickten linken Ventrikel mit erhaltener systolischer Funktion und schwerer diastolischer Dysfunktion mit restriktivem Füllungsmuster. Im Herz-MRI wurde eine diffuse, teils transmurale Fibrose beider Ventrikel und Vorhöfe dargestellt (Abbildung 4). Nach dieser Standortbestimmung gehen wir von einem Mischphänotyp der HCM und RCM aus. Die Bildgebung ist in diesem Fall sehr suggestiv für eine kardiale Amyloidose. Für die weitere Differentialdiagnose der Amyloidose wurden weitere Laboruntersuchungen (Immunfixation Serum und Urin) und eine weitere Bildgebung (99mTc-DPD- Szintigraphie) veranlasst. Infolge fehlender Hinweise für eine Plasmazelldyskrasie und Nachweis einer ausgeprägten Radionukleidanreicherung im linksventrikulären Myokard konnte die Diagnose einer Transthyretin Amyloidose gestellt werden. Aufgrund des sehr frühen Krankheitsauftretens erfolgte auch eine bioptische Sicherung der Diagnose mittels Endomyokardbiopsie. Nach einer genetischen Testung konnte eine genetische Form der Transthyretin Amyloidose ausgeschlossen werden, was wiederum für die Familie wichtig ist. Die genaue Stellung der Diagnose ermöglichte den Beginn einer Transthyretin-stabilisierenden Therapie.
Wie in diesem Beispiel demonstriert, führt die initiale Standortbestimmung zur Identifizierung des KMP-Phänotyps und markiert den Ausgangspunkt eines diagnostischen Prozesses, dessen Hauptziel darin besteht, die zugrunde liegende Ursache zu ermitteln. Die Erlangung einer ätiologischen Diagnose wird zunehmend relevant dank der Entwicklung neuer massgeschneiderter Behandlungen.
Die Bausteine der Differentialdiagnose umfassen nebst den klinischen Befunden eine multimodale Bildgebung, spezifische Laboruntersuchungen und Genetik.
Multimodale Bildgebung
Die multimodale Bildgebung bildet das Rückgrat für Diagnose und Verlaufskontrolle bei Patienten mit KMP. Die Echokardiographie ist die Methode der ersten Wahl für die Erfassung der kardialen Dimensionen, der Klappenfunktion und der systolischen/diastolischen Funktion. In den 2023 ESC KMP- Leitlinien wird die Rolle des kardialen MRI für die Gewebecharakterisierung unterstrichen und für alle KMP mit Klasse I, Evidenzgrad B, empfohlen. Im Falle der DCM ermöglicht das MRI eine Unterscheidung von der inflammatorischen DCM (11). Bei der HCM kann das MRI das Ausmass der myokardialen Fibrose zeigen mit direkter Beeinflussung der SCD-Risikostratifizierung und auch die Differenzierung von Amyloidose oder Morbus Fabry ermöglichen (12). Das MRI ermöglicht eine zuverlässige Evaluation der rechtsventrikulären Funktion bei Vorliegen einer arrhythmogenen rechtsventrikulären KMP (ARVC). Darüber hinaus ist das MRI in der Lage, die charakteristische fettige Ersatzfibrose direkt nachzuweisen. In seltenen Fällen von restriktiven KMP kann es beispielsweise mittels T1-T2- und T2*-Mapping die Sphingolipid-Akkumulation im Myokard bei Morbus Fabry oder eine Eisenüberladung bei Hämochromatose identifizieren (13). Andere bildgebende Verfahren, einschließlich nuklearmedizinischer Techniken und CT, sind bei ausgewählten Patienten mit KMP angezeigt. Beispielsweise kann ein 18F-FDG-PET nützlich zur Identifizierung einer aktiven Sarkoidose sein (14). Die DPD-Knochenszintigraphie kann zur Ätiologiebestimmung der Amyloidose helfen (15). Es ist insgesamt wichtig, dass Ärzte stets das Verhältnis von behandlungsrelevanten Resultaten zu den Vorteilen und Einschränkungen jeder Bildgebungstechnik abwägen.
Labordiagnostik
Eine umfassende Laboruntersuchung gehört zur Standortbestimmung bei Patienten mit KMP. Ein Differentialblutbild, Parameter zur Nieren- und Leberfunktion, Elektrolyte, Schilddrüsenfunktion sowie HbA1c sind für alle Patienten mit Symptomen einer Herzinsuffizienz empfohlen. Das NTproBNP und das high-sensitivity Troponin können für Diagnostik, Prognose und Therapiemonitoring nützlich sein. In Abhängigkeit der Verdachtsdiagnose kommen gezielte Laboruntersuchungen zum Einsatz, z. B. CRP bei Myokarditis, Ferritin- und Transferrinsättigung bei Hämochromatose, Creatinin-Kinase und Myoglobin bei Myopathien oder neuromuskulären Erkrankungen. Bei Hinweisen für seltenere metabolische oder syndromale Erkrankungen sollte eine Zuweisung in die spezialisierte Klinik für weitere gezielte Diagnostik erfolgen.
Genetik / Familienscreening
Generell gilt es, bei KMP zu betonen, dass häufig eine familiäre Komponente vorliegt und der zentrale Ausgangspunkt eine detaillierte Familienanamnese über mindestens 3 Generationen darstellt. Des Weiteren wird den erstgradigen Familienangehörigen ein klinisches Familienscreening mit EKG und Echokardiographie empfohlen. Sollten Rhythmusstörungen eine vordringliche Rolle spielen, ist in der Regel auch ein Langzeit-EKG empfohlen. Bei KMP, die v. a. mit strukturellen Veränderungen im Herz-MRI einhergehen (z. B. Laminopathie, Phospholamban oder Desmoplakin KMP), kann auch ein MRI im Rahmen des Familienscreenings erwogen werden (16, 17). Wichtig ist, dass das klinische Screening in regelmässigen Abständen wiederholt werden sollte, da sich die Krankheiten in praktisch jedem Lebensalter klinisch manifestieren können.
Ein genetisches Testen sollte prinzipiell bei allen Patienten mit KMP erwogen werden und pragmatisch in Abhängigkeit der therapeutischen Konsequenz und Familienanamnese auch durchgeführt werden. In der Tabelle 1 sind exemplarische Szenarien für die Indikationsstellung eines genetischen Tests bei Indexpatient/-innen und Angehörigen dargestellt. Dabei sollten auch ökonomische Überlegungen eine Rolle spielen. Beispielsweise hat eine genetische Untersuchung bei einem betagten Patienten mit einer apikalen Form der HCM ohne erstgradige Verwandten keine Konsequenz auf das therapeutische Management oder Kaskadenscreening.
Die identifizierten Mutationen werden entsprechend ihrer Pathogenizität in 5 Kategorien eingeteilt: gutartig (Klasse 1), wahrscheinlich gutartig (Klasse 2), Variante unklarer Signifikanz (Klasse 3) wahrscheinlich pathogen (Klasse 4), pathogen (Klasse 5) (18). Da die Stärke des Phänotyps die Wahrscheinlichkeit der Pathogenizität beeinflusst, sollte der klinische Phänotyp in die Interpretation der Varianten integriert werden. Entscheidungen, ob eine Variante unklarer Signifikanz mit der Krankheit assoziiert werden kann, sollten von Fall zu Fall und in enger Zusammenarbeit mit Experten in Kardiogenetik und KMP im Rahmen einer interdisziplinären Teamdiskussion getroffen werden.
Nicht alle Personen, die eine Mutation tragen, manifestieren die Krankheit auch tatsächlich klinisch (unvollständige Penetranz), und bei denen, die dies tun, gibt es eine breite Variabilität bezüglich Alter des Auftretens und Schweregrad der Erkrankung (19). Bei Kindern sollte unbedingt auf eine Mitsprachemöglichkeit geachtet werden. Genetische Testung im Rahmen eines Kaskadenscreenings ist bei Kindern besonders empfohlen, wenn die KMP bei der betroffenen Indexperson im Kindesalter aufgetreten ist. Wenn die Kinder kompetitiven Sport betreiben oder einen Beruf anstreben, welcher bei Ausbildung einer KMP nicht längerfristig ausgeübt werden kann, sollte ebenfalls im Kindesalter eine genetische Testung erwogen werden.
Zu betonen ist, dass sich die Klassifizierung der Mutation in den untersuchten Genen im Laufe der Zeit ändern kann. Daher ist es entscheidend für den Kliniker, die Richtigkeit der Pathogenizitätszuweisung für jede identifizierte Variante erneut zu überprüfen, anstatt sich ausschliesslich auf die Laborinterpretation zu verlassen. Wichtig ist hier die Zusammenarbeit mit genetischen Fachärzten, die über das entsprechende Wissen im Bereich der Kardiogenetik verfügen. Die Rolle der Kardiogenetik-Fachperson ist nicht nur für eine umfassende Prä- und Post-Testberatung, sondern auch für die Interpretation der Resultate zentral. In Anbetracht der sich stets wandelnden Datenlage erfolgt eine Überprüfung der vorliegenden genetischen Resultate oder eine Wiederholung der genetischen Testung am besten in Absprache mit einer KMP- Klinik, die über die entsprechende Expertise verfügt (20).
Empfehlung zu Management der Patienten mit KMP
Nach Erreichen der spezifischen Diagnose ist es entscheidend, bestimmte therapeutische Massnahmen zu ergreifen, entweder um klinisch relevante Arrhythmien rechtzeitig zu erkennen, den SCD zu vermeiden oder im besten Fall sogar den Krankheitsverlauf modifizieren zu können. Die Zusammenarbeit mit KMP Kliniken kann eine umfassende und multidisziplinäre Behandlung der Patienten und ihren Angehörigen anbieten.
Rhythmusstörungen
Regelmässige EKG-Untersuchungen (12-Ableitungs-EKG und Langzeit-EKGs) sind bei der initialen klinischen Evaluation und in regelmässigen Abständen nützlich, um das SCD- Risiko abschätzen zu können und sowie ein frühzeitiges Erkennen von Vorhofflimmern zu ermöglichen mit dem Ziel, Schlaganfälle zu verhindern. Das 12-Ableitungs-Ruhe-EKG ist häufig die erste Untersuchung, die auf eine KMP hinweisen kann. Obwohl das EKG oft unspezifisch ist, gibt es bestimmte Merkmale wie atrioventrikuläre Blockbilder (z. B. bei Sarkoidose/Laminopathie/Amyloidose), ventrikuläre Präexzitation (z. B. bei Morbus Fabry/PRKAG2-KMP sowie andere Glykogenspeicherkrankheiten), Repolarisationsstörungen (z. B. bei ARVC/HCM) sowie hohe oder niedrige QRS-Amplituden (z. B. bei HCM/Phospholamban-MP/Glykogenspeicherkrankheiten/Amyloidose), die auf eine bestimmte Ätiologie hinweisen können.
Vorhofflimmern ist die häufigste Arrhythmie in allen Untergruppen der KMP. Insbesondere bei HCM und RCM ist das Vorhofflimmern mit erhöhtem Schlaganfallrisiko assoziiert. Folglich wird bei diesen Phänotypen bei Vorliegen eines Vorhofflimmerns unabhängig vom CHA2DS2-Vasc-Score eine therapeutische Antikoagulation empfohlen (21). Es werden nun seit Längerem die neuen Antikoagulanzien als Erstlinientherapie empfohlen.
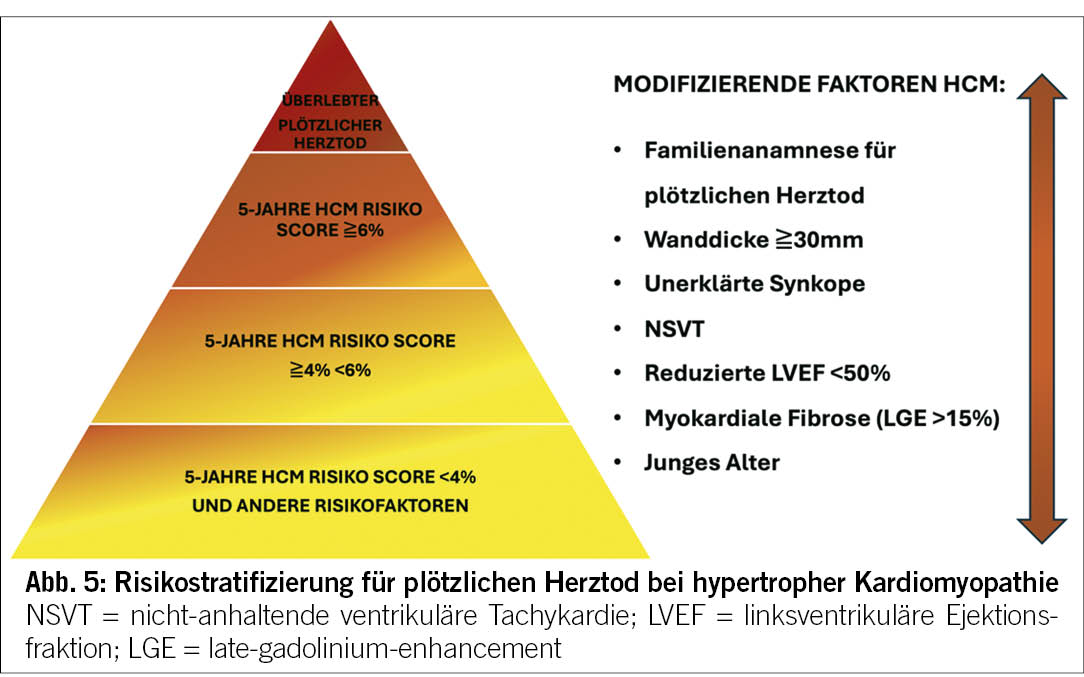
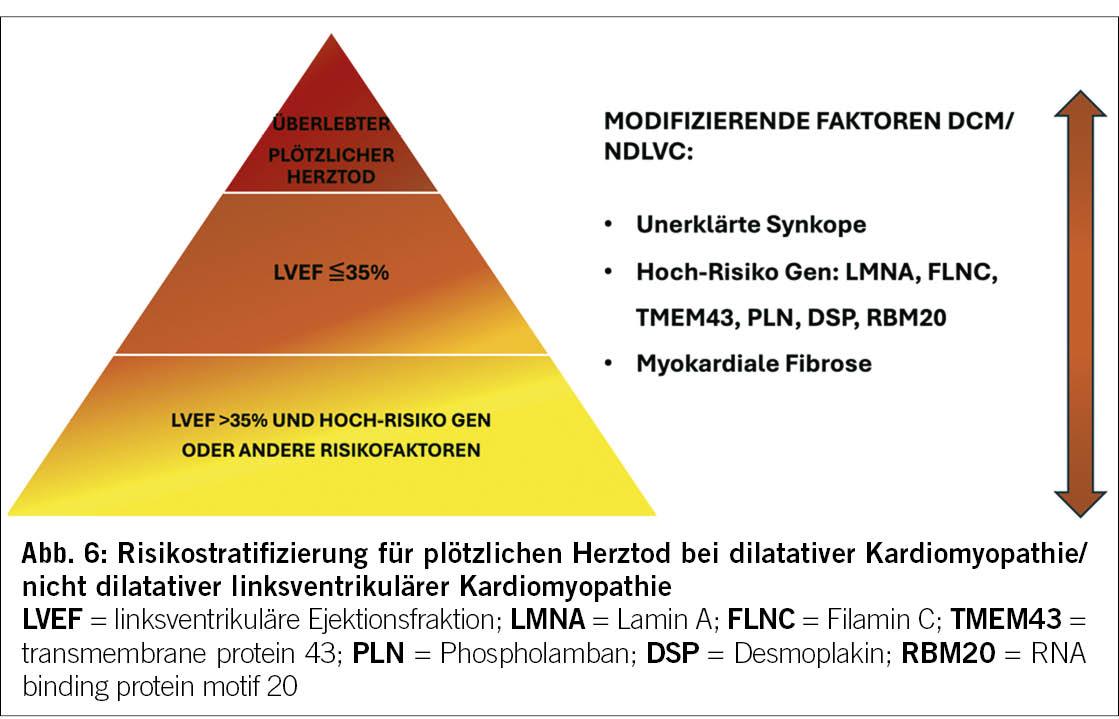
Die 2023 ESC KMP-Richtlinien haben die Empfehlungen bezüglich der Risikostratifizierung des SCD in den verschiedenen Phänotypen aktualisiert. Die Hauptinnovation liegt im Stellenwert der Genetik und des Ausmasses der myokardialen Fibrose quantifiziert im Herz-MRI bei der Risikoeinschätzung des SCD.
Der seit 2014 in den ESC HCM-Behandlungsrichtlinien implementierte HCM-Risk-SCD-Kalkulator spielt auch aktuell immer noch eine zentrale Rolle in der Risikoeinschätzung (22). Neu gelten aber auch die Anwesenheit von LGE (>15%) und eine linksventrikuläre Auswurffraktion von <50% als modifizierende Faktoren bei Patienten mit HCM und niedrig-mässigem Risiko für plötzlichen Herztod (Abbildung 5). Patienten mit NDLVC haben in der Regel eine normale oder leicht reduzierte LVEF. Demzufolge ist bei diesen Patienten der Genotyp der ausschlaggebende Faktor zur Bestimmung des SCD-Risikos (Abbildung 6). Im Bereich der NDLVC und DCM sind Mutationen in folgenden Genen bereits bei einer LVEF >= 35% mit einem erhöhten SCD-Risiko verbunden: Desmoplakin (DSP), transmembrane protein 43(TMEM43), Filamin C (FLNC), Lamin A (LMNA), Phospholamban (PLN) und RNA binding protein motif 20 (RBM20).
Medikamentöse Therapie
Die medikamentösen Therapien können in 3 Gruppen eingeteilt werden:
A. Stehen ursächliche Therapien zur Verfügung?
B. Behandlung der Obstruktion des linksventrikulären Ausflusstrakts bei HCM
C. Therapie der Herzinsuffizienz
Ad A.: Krankheitsmodifizierende therapeutische Massnahmen sind verfügbar für bestimmte KMP, z. B. die Enzymersatztherapie oder die Chaperone-Therapie für den Morbus Fabry und die Protein-Transthyretin-Stabilisatoren l für Transthyretin-assoziierte Amyloidose (23, 24).
Ad B.: Eine Neuigkeit der 2023 ESC KMP-Leitlinien ist die Empfehlung für Mavacamten als Zweitlinientherapie (IIa) bei Patienten mit HCM und symptomatischer LVOT-Obstruktion bei insuffizienter oder nicht tolerierter medikamentöser Therapie mit Betablockern, Calciumantagonisten und/oder Disopyramid. Dabei handelt es sich um einen oral verfügbaren allosterischen Inhibitor der kardialen Myosin-Adenosintriphosphatase (ATPase), der durch die Reduktion der Bildung von Aktin-Myosin-Querbrücken die exzessive myokardiale Kontraktilität verringert und die diastolische ventrikuläre Füllung verbessert. Erste randomisierte Studien haben die Wirksamkeit dieser Therapie gezeigt (25, 26). Aufgrund der negativen Inotropie ist eine engmaschige und regelmässige Kontrolle der LVEF vonnöten, bis entsprechende Langzeitdaten vorliegen. Es bleibt zudem abzuwarten, ob Myosin-Inhibitoren auch krankheitsmodifizierend bei der nicht obstruktiven HCM wirken.
Ad C.: Die Behandlung der Herzinsuffizienz ist primär abhängig von der linksventrikulären systolischen Funktion und den Symptomen/NYHA-Klasse (27). Daher sind diese Empfehlungen für Patienten mit Herzinsuffizienz mit reduzierter linksventrikulärer Auswurffraktion (HFrEF) und spielen bei allen KMP eine wichtige Rolle. Hier ist zentral, dass bei der HCM bereits eine LVEF <50% als Phase der systolischen Herzinsuffizienz gilt, da diese Patienten infolge der Hyperkontraktilität mit abnorm hohen LVEFs >70% starten und daher bereits eine LVEF <50% als substanziell erniedrigt gilt (28). Empfehlungen für das Management von Herzinsuffizienz mit erhaltener Auswurffraktion (HFpEF) sind hauptsächlich auf die nicht obstruktive HCM und die RCM anwendbar. Besonders die SGLT2-Hemmer haben sich nicht nur als wichtige Säule der Therapie bei HFrEF, sondern auch als erste Medikamentenklasse mit Verbesserung des kardiovaskulären Outcomes bei HFpEF etabliert (29, 30).
Warum und wann in eine KMP Klinik zuweisen?
KMP haben viele Facetten, angefangen bei der Diagnostik bis hin zu Risikostratifizierung und den verschieden therapeutischen Aspekten, was die Beteiligung verschiedener Disziplinen (Imaging, klinische Kardiologie, Genetik, Rhythmologie, Herzchirurgie etc.) mit sich bringt. Diese Anforderungen werden idealerweise in den darauf spezialisierten Zentren mit multidisziplinären Teams erfüllt. Interventionelle Verfahren (z. B. Septalalkoholablationen, chirurgische Myektomien, etc.) erfordern eine Expertise, die nur Zentren mit hohen Fallzahlen erreichen können.
Wir denken daher, dass bei komplexen Patienten mit z. B. KMP unklarer Ätiologie, hohem oder unklarem Risiko für klinische Komplikationen oder Bedarf für Therapieausbau eine Zuweisung in eine KMP-Klinik sinnvoll ist. Zudem kann auch eine einmalige Standortbestimmung in einer KMP-Klinik helfen, die aktuellen Therapieoptionen unter permanenter wissenschaftlicher Aktualisierung zu überprüfen.
Das Ziel der Zuweisung in eine KMP-Klinik ist eine umfassende Betreuung der Patienten/-innen und deren Angehörigen im Sinne einer guten Zusammenarbeit und einer «shared care» mit den Zuweisern.
Klinik für Kardiologie
Universitätsspital Zürich
Rämistrasse 100
8091 Zürich
Die Autorinnen haben keine Interessenkonflikte im Zusammenhang mit diesem Artikel deklariert.
Literatur
1. Elliott, P., et al., Classification of the cardiomyopathies: a position statement from the european society of cardiology working group on myocardial and pericardial diseases. European Heart Journal, 2007. 29(2): p. 270-276.
2. Teare, D., Asymmetrical hypertrophy of the heart in young adults. Br Heart J, 1958. 20(1): p. 1-8.
3. Morrow, A.G. and E.C. Brockenbrough, Surgical treatment of idiopathic hypertrophic subaortic stenosis: technic and hemodynamic results of subaortic ventriculomyotomy. Ann Surg, 1961. 154(2): p. 181-9.
4. Arbelo, E., et al., 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J, 2023. 44(37): p. 3503-3626.
5. Rapezzi, C., et al., Diagnostic work-up in cardiomyopathies: bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. European Heart Journal, 2012. 34(19): p. 1448-1458.
6. Marcus, F.I., et al., Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J, 2010. 31(7): p. 806-14.
7. Faber, J.W., et al., Lack of morphometric evidence for ventricular compaction in humans. J Cardiol, 2021. 78(5): p. 397-405.
8. Petersen, S.E., et al., Excessive Trabeculation of the Left Ventricle: JACC: Cardiovascular Imaging Expert Panel Paper. JACC Cardiovasc Imaging, 2023. 16(3): p. 408-425.
9. Arbustini, E., et al., Left Ventricular Noncompaction: A Distinct Genetic Cardiomyopathy? J Am Coll Cardiol, 2016. 68(9): p. 949-66.
10. Romero Puche, A.J., et al., Mixed phenotypes: implications in family screening of inherited cardiomyopathies. European Heart Journal, 2013. 34(suppl_1).
11. Ferreira, V.M., et al., Cardiovascular Magnetic Resonance in Nonischemic Myocardial Inflammation: Expert Recommendations. J Am Coll Cardiol, 2018. 72(24): p. 3158-3176.
12. Maron, B.J., et al., Diagnosis and Evaluation of Hypertrophic Cardiomyopathy. Journal of the American College of Cardiology, 2022. 79(4): p. 372-389.
13. Messroghli, D.R., et al., Clinical recommendations for cardiovascular magnetic resonance mapping of T1, T2, T2* and extracellular volume: A consensus statement by the Society for Cardiovascular Magnetic Resonance (SCMR) endorsed by the European Association for Cardiovascular Imaging (EACVI). J Cardiovasc Magn Reson, 2017. 19(1): p. 75.
14. Chareonthaitawee, P., et al., Joint SNMMI-ASNC Expert Consensus Document on the Role of (18)F-FDG PET/CT in Cardiac Sarcoid Detection and Therapy Monitoring. J Nucl Med, 2017. 58(8): p. 1341-1353.
15. Gillmore, J.D., et al., Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation, 2016. 133(24): p. 2404-12.
16. Te Rijdt, W.P., et al., Myocardial fibrosis as an early feature in phospholamban p.Arg14del mutation carriers: phenotypic insights from cardiovascular magnetic resonance imaging. Eur Heart J Cardiovasc Imaging, 2019. 20(1): p. 92-100.
17. de Frutos, F., et al., Late gadolinium enhancement distribution patterns in non-ischaemic dilated cardiomyopathy: genotype-phenotype correlation. Eur Heart J Cardiovasc Imaging, 2023. 25(1): p. 75-85.
18. Richards, S., et al., Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 2015. 17(5): p. 405-24.
19. Topriceanu, C.C., et al., Meta-Analysis of Penetrance and Systematic Review on Transition to Disease in Genetic Hypertrophic Cardiomyopathy. Circulation, 2024. 149(2): p. 107-123.
20. Ahmad, F., et al., Establishment of Specialized Clinical Cardiovascular Genetics Programs: Recognizing the Need and Meeting Standards: A Scientific Statement From the American Heart Association. Circ Genom Precis Med, 2019. 12(6): p. e000054.
21. Hindricks, G., et al., 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur Heart J, 2021. 42(5): p. 373-498.
22. O‘Mahony, C., et al., A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J, 2014. 35(30): p. 2010-20.
23. Garcia-Pavia, P., et al., Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J, 2021. 42(16): p. 1554-1568.
24. Linhart, A., et al., An expert consensus document on the management of cardiovascular manifestations of Fabry disease. Eur J Heart Fail, 2020. 22(7): p. 1076-1096.
25. Desai, M.Y., et al., Mavacamten in Patients With Hypertrophic Cardiomyopathy Referred for Septal Reduction: Week 56 Results From the VALOR-HCM Randomized Clinical Trial. JAMA Cardiol, 2023. 8(10): p. 968-977.
26. Olivotto, I., et al., Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet, 2020. 396(10253): p. 759-769.
27. McDonagh, T.A., et al., 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) With the special contribution of the Heart Failure Association (HFA) of the ESC. European Heart Journal, 2021. 42(36): p. 3599-3726.
28. Marstrand, P., et al., Hypertrophic Cardiomyopathy With Left Ventricular Systolic Dysfunction: Insights From the SHaRe Registry. Circulation, 2020. 141(17): p. 1371-1383.
29. Anker, S.D., et al., Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N Engl J Med, 2021. 385(16): p. 1451-1461.
30. Solomon, S.D., et al., Dapagliflozin in Heart Failure with Mildly Reduced or Preserved Ejection Fraction. N Engl J Med, 2022. 387(12): p. 1089-1098.