Eine 52-jährige Frau, mit seit 2007 bekannter Sklerodermie, welche sich durch typisches Raynaud Phänomen, Sklerodactylie, Teleangiektasien und Störung der Oesophagusmotilität manifestierte, wurde wegen zunehmender Anstrengungsdyspnoe zugewiesen. Klinisch zeigten sich Teleangiektasien am Dekolleté und eine Sklerodactylie. Die Bodyplethysmographie war unauffällig, die CO-Diffusionskapazität war leicht reduziert. Die Sechs-Minuten-Gehstrecke war ebenfalls leicht reduziert (430 Meter) mit Entsättigung von 97 auf 91%. Eine transthorakale Echokardiografie zeigte eine erhöhte maximale Geschwindigkeit der Trikuspidalinsuffizienz von 3,2 m/s, woraus sich ein Druckgradient von 40 mmHg errechnet. Es findet sich ein normaler rechter Ventrikel mit normaler Funktion. Welche Untersuchung ist als nächstes angezeigt? [5].
Einleitung
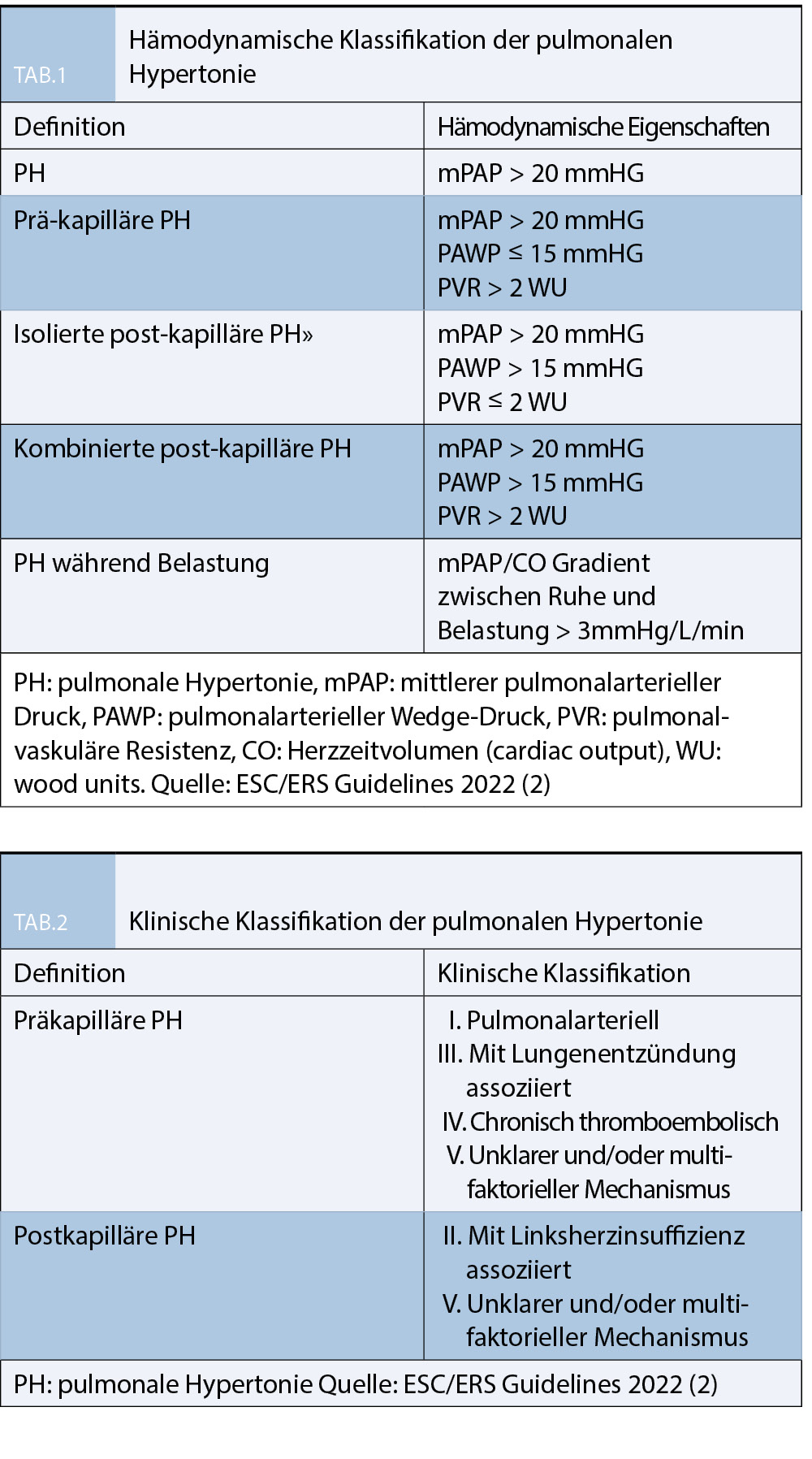
Die pulmonale Hypertonie (PH) umfasst eine Reihe lebensverkürzenden Erkrankungen, die durch einen erhöhten mittleren pulmonalarteriellen Druck (mPAP) > 20 mmHg gekennzeichnet sind und die unbehandelt zu einer zunehmenden Belastungsdyspnoe, Rechtsherzversagen und Tod führen können (6). Die Erhöhung des mPAP entwickelt sich entweder im Rahmen einer pulmonalen Vaskulopathie (WHO Gruppe 1 und 4, pulmonal-arterielle und chronisch-thromboembolische PH), beide gekennzeichnet durch einen erhöhten pulmonalvaskulären Widerstand (PVR) oder auf dem Boden einer chronischen Linksherzinsuffizienz oder Klappenvitien, gekennzeichnet durch einen erhöhten pulmonal-arteriellen Verschlussdruck (Wedge-Druck oder PAWP) oder bei chronischen Lungenkrankheiten durch die hypoxische pulmonale Vasokonstriktion und Rarefizierung des Lungengefässbettes (7). Bereits am ersten Weltsymposium 1973 in Genf wurde die PH als „während einer Rechtsherzkatheteruntersuchung dokumentierte Erhöhung des mPAP auf mindestens 25 mmHg“ definiert. Diese Definition wurde im Wesentlichen über Jahrzehnte beibehalten, und erst in den im August 2022 neu erschienenen Guidelines der Europäischen Kardiologischen und Respiratorischen Gesellschaft aufgrund neuer Erkenntnisse der normalen Drücke und
Widerstände im Lungenkreislauf auf den Grenzwert mPAP >20 mmHg und PVR >2 WU angepasst (2). Der pulmonalarterielle Druck wird durch das Herzminutenvolumen (cardiac output, CO), den PVR und PAWP beeinflusst. Es ist für die Behandelnden entsprechend wichtig, diese Faktoren zu kennen (8). Aus den gemessenen Grössen kann der PVR einfach berechnet werden:

Hämodynamische Klassifikation
Zur Diagnose und Einteilung der PH ist es angebracht,
zwischen prä- und post-kapillärer PH zu unterscheiden, hierzu dient wie in Tabelle 1 ersichtlich der PAWP und der PVR, welcher sich als mPAP-PAWP/cardiac output (CO) berechnet. Für diese Berechnung braucht es den CO, welcher zwingend mittels direktem Fick, d.h. unter Messung der aktuellen Sauerstoffaufnahme oder mittels Thermodilution erfolgen soll. Der indirekte Fick-Test oder andere nicht-invasive Methoden sind obsolet. Da der PAWP je nach Volumenstatus des Patienten jedoch auch deutlich variieren kann, empfiehlt es sich, für die Unterscheidung prä- und postkapillär auch die klinische Wahrscheinlichkeit mit einzubeziehen. Alter > 70 Jahre, Adipositas, Hypertonie, Dyslipidämie, Diabetes oder Glukose-Intoleranz, Vorhofflimmern, bekannte Kardiopathie, Linksschenkelblock, linksventrikuläre Hypertrophie im EKG oder im TTE, Dilatation des linken Vorhofs sind Faktoren, die die Wahrscheinlichkeit einer post-kapillären PH stark erhöhen.
Die neue hämodynamische Definition beinhaltet auch die PH unter Belastung. Bei dieser Messung wird die komplette pulmonale Hämodynamik unter Fahrradergometerbelastung mit Stufenprotokoll am Ende jeder 2-3-minütigen Stufe gemessen, empfohlen wird dabei den CO mittels direkter Fick-Methode unter Einbezug des VO2-Messung zu ermitteln (9).
Aus hämodynamischer Sicht kann die pulmonale Hypertonie in 3 Gruppen aufgeteilt werden: prä-kapilläre, isoliert post-kapilläre und kombiniert prä- und post-kapilläre pulmonale Hypertonie (Tabelle 1).
Ein PAWP ≤15 mmHg definiert eine prä-kapilläre PH, ein PAWP > 15 mmHg eine post-kapilläre pulmonale Hypertonie. Ein PVR von > 2 Wood units (WU) ist bereits pathologisch und deutet auf eine Vaskulopathie hin und wird als Grenzwert definiert. Die prä-kapilläre PH zeigt sich mit einem PVR von > 2 WU, die post-kapilläre PH mit einem PVR ≤ 2 WU.
Klinische Klassifikation
Aus dieser hämodynamischen Klassifikation und der entsprechenden Klinik wird die pulmonale Hypertonie in fünf verschiedene Gruppen aufgeteilt (Tabelle 2) (2). Gruppe 1 wird als pulmonal-arterielle Hypertonie (PAH) bezeichnet und gehört zusammen mit der Gruppe 4 chronisch-thromboembolische PH zu den pulmonal-vaskulären Krankheiten, definiert durch eine präkapilläre PH und eine Pathologie in den Pulmonalgefässen. Die PAH kann idiopathisch (IPAH) oder hereditär sein. Bei der IPAH ist insbesondere die Abgrenzung der vasoreaktiven Form wichtig, da diese für Patienten erhebliche therapeutische Konsequenzen hat. Die Vasoreaktivitätstestung mit NO (Nitrogenmonoxid), inhaliertem Iloprost oder Epoprostenol intravenös gehört daher zu jeder diagnostischen Rechtsherzkatheteruntersuchung der PAH. Diverse Drogen und Toxine können eine PAH ebenfalls verursachen. Die PAH ist ebenso mit gewissen Erkrankungen assoziiert, wie Kollagenose (v.a. Sklerodermie), portopulmonale Hypertonie bei einer portalen Hypertonie, HIV-Infektion, kongenitale Herzerkrankungen oder Schistosomiasis. Des Weiteren gehören zu dieser Gruppe die seltenen pulmonale venookklusive Krankheit, die pulmonale kapilläre Hämangiomatose und die persistierende pulmonale Hypertonie den Neugeborenen. Die PAH ist eine seltene Krankheit (Inzidenz 6/Million und Prävalenz 48-55 Fälle/Million) (2). Die idiopathische Form ist der häufigste Subtyp (50-60%), gefolgt von der PAH assoziiert mit einer Kollagenose, kongenitalen Herzerkrankungen und portaler Hypertonie. Insbesondere bei der Sklerodermie kann die Rechtsherzkatheteruntersuchung mit Belastungsprotokoll eine wichtige Hilfe zur Frühdiagnose sein.
Die Gruppe 2 umfasst die PH bei Linksherzerkrankungen mit erhaltener oder reduzierter Ejektionsfraktion oder bei Klappenvitien. Es ist daher meistens eine post-kapilläre PH, seltener eine kombinierte prä- und post-kapilläre PH vorhanden. Die PH Gruppe 2 ist die häufigste Ursache für eine PH (70%) und ist bei Patienten mit Herzinsuffizienz sehr häufig (circa 50%), noch häufiger bei Patienten mit schwerer Erkrankung der Mitralklappen (60-70%) (2).
Die Gruppe 3 ist eine prä-kapilläre PH, die sich bei pulmonalen Erkrankungen und/oder Hypoxämie findet. Eine schwere PH ist bei 1-5% der Patienten mit schwerer COPD und bei 30-50 % der Patienten mit schwerer idiopathischer Lungenfibrose zu finden und verschlechtert die Prognose diesen Krankheiten erheblich (2).
Die vierte Gruppe stellt die chronisch-thromboembolische PH (CTEPH) dar und hat ebenfalls eine prä-kapilläre Ursache. Die Inzidenz und Prävalenz sind in den Registerdaten tief (2-6/Million und 26-38 Fälle/Million). Sie wird jedoch vermutlich deutlich unterschätzt und zeigt im klinischen Alltag dank besserer Erkenntnis und Screening eine kontinuierliche Zunahme (2).
Weitere metabolische, hämatologische oder systemische Erkrankungen (z.B. Sarkoidose, pulmonale Langerhanszell Histiozytose), die eine pulmonale Hypertonie verursachen können gehören zur fünften Gruppe (3), hämodynamisch gesehen können die verursachenden Entitäten prä-, post-kapillär oder kombiniert sein.
Genetische Aspekte
Genetische Aberrationen haben hauptsächlich in der ersten Gruppe der pulmonalen Hypertonie einen hohen Stellenwert. Die Mutation vom Gen BMRP 2 wurde in 75% der familiären, und 25% der sporadischen pulmonal-arteriellen Hypertonie gezeigt (2). Mutationen in den Genen vom transformierenden Wachstumsfaktor Beta (TGF Beta) wurden in Patienten mit pulmonal-arteriellen Hypertonie nachgewiesen. Bezüglich der Gruppe 3 ist interessant festzustellen, dass die Ausprägung der pulmonalen Hypertonie in hypoxämischen COPD-Patienten durch Genpolymorphismus im Serotonin Tansporter Gen (5-HTT) beeinflusst wird (10) (11).
Pathologie
Bei der PAH und sekundär auch bei gewissen anderen PH-Formen weisen die distalen-muskulären Arterien in allen Wandschichten Zellproliferationen auf, welche zur Wandverdichtungen führen. Insbesondere bei den Gefässverzweigungen finden sich zum Teil auch «krebsähnliche» Zellwandwucherungen, die sogenannten plexiformen Läsionen (11). Hierdurch kommt es zu eingeengtem Gefässlumen, Dilatation der vorangehenden Gefässabschnitte und zu einem erhöhten PVR. Pathologisch sind häufig neben den präkapilläre Arteriolen auch Kapillaren sowie die postkapilläre Venolen, so dass heute von einem Kontinuum der PAH bis zur venookklusiven Krankheit ausgeht. Durch eine verminderte BMPR2-Expression in den pulmonalen Endothelzellen verändert sich die lokale Zytokinproduktion. Zum Beispiel, der Spiegel vom Interleukin 17A und Interleukin 6 erhöht sich (16). Durch den Wachstumsfaktor Granulocyte-Macrophage Colony Stimulating Factor (GM-CSF) werden Makrophagen rekrutiert und eine Endotheladhäsion bildet sich. Die Gefässe werden plexiform, ähnlich einer Zwiebel umgebaut. Sie nehmen an Gesamtdurchmesser zu, das Gefässlumen engt sich ein (11). Die plexiformen Läsionen sind um und in der Adventitia, in der Media oder in der Intima lokalisiert. In der Adventitia und im peribronchialen Bindegewebe sind Gefässe des systemischen Kreislaufes, wie die vasa vasorum und die Bronchialarterien eingebettet. Diese Gefässe scheinen durch Mikrogefässanastomosen mit den pulmonalen Arterien zu kommunizieren und eine Shuntfunktion zu erfüllen. Durch die BMPR2-Mutation tritt eine Hypertrophie und Erweiterung der Bronchialarterien und eine Vermehrung dieser bronchialen Mikrogefässen auf. Auch grössere Gefässstrukturen, sogenannte singular millimetric fibrovascular lesions (SiMFis) verbinden die Bronchialarterien und die pulmonalen Arterien und Venen.
Bei der CTEPH finden sich Gefässwandvernarbungen nach stattgehabten, pathologisch nicht abgebauten Thromboembolien oder in-situ Thrombosen. Durch den dadurch erhöhten PAP entwickeln sich sekundär in den Arteriolen ähnliche Veränderungen wie bei der PAH, auch hier können die Veränderungen über die Arteriolen, Kapillaren bis in die Venolen gehen.
Die chronische Hypoxie und auch Hypoxämie spielt insbesondere für Gruppe 3 PH, jedoch auch bei anderen PH-Formen, eine entscheidende Rolle. Die akute pulmonale Vasokonstriktion unter Hypoxie ist ein physiologischer Regulationsmechanismus im pulmonalen Kreislauf, der das Ventilations-/Perfusionsverhältnis optimiert und die suffiziente Oxygenierung des Blutes gewährleistet (12). Aufgrund einer Vermehrung der Hypoxiesensorproteine HIF1 und HIF2 (13) wird eine Vasokonstriktion bei geringer Hypoxie ausgelöst, diese ist gemessen an den aktuell herrschenden Bedingungen verfrüht. Es entsteht ein Teufelskreis.
Auch eine gewisse Maladaptation des Immunsystems trägt zum vaskulären Remodelling bei (13). Ionenkanäle sind von wesentlicher Bedeutung in der Regulierung des Vasotonus (12). Bei einer gestörten Funktion von Kaliumkanälen erhöht sich die intrazelluläre Kaliumkonzentration und kommt eine Vasokonstriktion auf. Die Mutationen von TASK 1 Kaliumkanal (TWIK-related acid-sensitive
potassium channel 1) sind in Patienten mit idiopathischer pulmonalen Hypertonie beschrieben (12).
Durch die Endothelverletzung kommt die glatte Gefässmuskulatur mit Wachstumsfaktoren und Mitogenen des Blutes in Kontakt. Die folgenden drei Signalisationswege sind in der Entstehung wichtig: Prostacyclin, Nitrogenmonoxid/ Cyclic Guanoside Monophosphate und der Endothelin
Pathway. Es entsteht ein Ungleichgewicht zwischen den vasokonstriktiven und proproliferativen Endothelin und den vasodilatativen und antiproliferativen Nitrogenmonoxid und Prostacyclin Signalisationsweg. Die Thrombozyten haben eine wichtige Rolle in der Entstehung der prokoagulatorischen Effekt, da sie Serotonin, Vaskular Endothelial Growth Faktor und Platelet-derived Growth Faktor produzieren (14).
Pathophysiologie
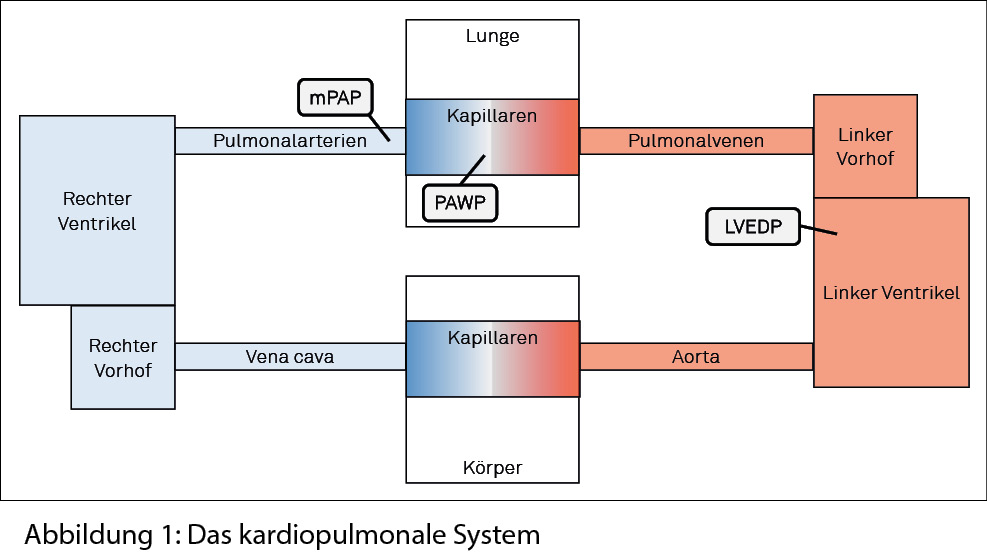
Die rechtsventrikuläre Funktion ist der fundamentale Faktor in der Prognose einer schweren PH. Da die Rechtsherzinsuffizienz sich in der pulmonalen Hypertonie durch die erhöhte Nachlast und das dadurch gestörte ventrikulo-
arterielle Coupling entwickelt, ist das pathophysiologische Verständnis des kardiopulmonalen Systems sehr wichtig (Abbildung 1).
Im Hinblick auf die PH sind der rechte Ventrikel und das pulmonale Gefässystem, insbesondere die kleinen Arterien bis zu den Kapillaren von grosser Bedeutung. Intrinsische Faktoren, die die Rechtsherzfunktion beeinflussen sind die Kontraktilität, die myokardiale Steifigkeit und die Dauer der ventrikulären Relaxation (15). Die intrinsischen Faktoren, sowie die Vor- und Nachlast bestimmen die globale rechtsventrikuläre Funktion. Die intrinsischen Faktoren werden in der Ejektionsfraktion, die Last in Drücke (mittlerer, systolischer, diastolischer) berücksichtigt. Im pulmonalen Kreislauf ist das Load teils permanent, insbesondere in den grossen Gefässen jedoch pulsierend. Da der Herzauswurf und somit der pulmonale Blutfluss unter Belastung sich deutlich erhöhen muss, ist der Fluss in den Pulmonalgefässen und Kapillaren ein sehr dynamischer Prozess, es braucht daher die Möglichkeit der Distension und des Recruitments von nur intermittierend durchbluteten Gefässen. Der Lungenkreislauf verfügt als Niederdrucksystem also über eine sehr hohe Anpassungsfähigkeit in den Kapillaren. Da dies ein dynamischer Prozess ist, werden sie nur intermittierend durchblutet. Falls der Fluss sich leicht zunimmt, erfolgt eine Vermehrung den pulmonalen Kapillaren. Falls es nicht mehr ausreicht, erfolgt deren Ausdehnung. Die Anpassungsfähigkeit, die sogenannte pulmonal-arterielle Compliance. Bei Zunahme des PVRs nimmt die pulmonal-arterielle Compliance ab, ebenso das ventrikulo-arterielle Coupling und es droht die Rechtsherzinsuffizienz. Demzufolge ist die pulmonal-arterielle Compliance grösser bei Patienten mit milder pulmonalvaskulärer Erkrankung, da dort auch die pulmonalvaskuläre Resistenz geringer ist.
Diagnose
Das Hauptsymptom eines Patienten/einer Patientin mit PH ist eine Anstrengungsdyspnoe. Hinzu kommen Fatigue und schnelle Erschöpfung, Dyspnoe beim vorne Beugen (sogenannte Bendopnoe), Thoraxschmerz und Palpitationen. Bei fortgeschrittenem Stadium stehen die Symptome einer Rechtsherzinsuffizienz: Unterschenkelödeme, abdominale Stauung, Aszites, Gewichtszunahme, Synkope im Vordergrund. Selten kann eine Hämoptyse oder Heiserkeit auf-
treten (16).
Im Status sind ein positiver hepatojugulärer Reflux, Unterschenkelödeme, Hepato- oder Splenomegalie oder Herzgeräusche aufgrund der pulmonalen Regurgitation typisch. Ein Elektrokardiogramm kann insbesondere zusammen mit dem BNP für das Screening benutzt werden: Sind beide normal, so ist das Vorliegen einer relevanten pulmonalen Drucksteigerung eher unwahrscheinlich. Ein Routinelabor inklusive Hämatogramm und Chemie mit Leberfunktions- und Schilddrüsenwerte gehört zum diagnostischen Workup. Typisch ist der BNP oder NT-proBNP-Wert aufgrund der Rechtsherzinsuffizienz erhöht. Eine Serologie mit antinuklearem Antikörper soll bei Verdacht auf Sklerodermie durchgeführt werden, sowie ein Hepatitis- und HIV-Screening. Die Lungenfunktionsprüfung zeigt meist normale Volumina – typisch ist jedoch eine leicht bis mittelgradig verminderte CO-Diffusionskapazität.
Die relevanteste Screeninguntersuchung stellt die transthorakale Echokardiographie dar, weist jedoch eine tiefe Sensitivität und Spezifität auf, da sie stark vom Untersucher abhängig ist (3). Mit der Geschwindigkeitsmessung der trikuspidalen Regurgitation kann der systolische pulmonalarterielle Druck abgeschätzt werden. Die Messung der rechtsventrikulären Dimension und Funktion ist ebenfalls wichtig sowie die Suche nach D-Shaping des linken Ventrikels und nach einem Perikarderguss.
Die Echokardiografie zeigt auch die linksventrikuläre Funktion, sowohl systolisch als diastolisch, und kann auf Klappenvitien oder intrakardialen Shunt hinweisen. Mittels Echokardiographie können der PAWP und der CO nur in Annäherungsformeln abgeschätzt werden (2). Beim Vorliegen eines erhöhten systolischen PAH ist eine PH möglich, dieser Wert ist jedoch nicht spezifisch. Es ist eine weitere Abklärung mittels Rechtsherzkatheteruntersuchung wie oben beschrieben empfohlen.
Beim Patienten mit einer idiopathischen, vererbbaren oder Medikament/Toxin induzierten pulmonalen Hypertonie sollte eine Vasoreaktivitätstestung durchgeführt werden. Während der Rechtsherzkatheteruntersuchung wird NO (Nitrogenmonoxid) oder Iloprost inhaliert und die Hämodynamik darunter gemessen. Der Test ist positiv bei Abnahme des mPAP um ≥ 10mmHg und bei einem absoluten mPAP von ≤ 40 mmHg bei konstantem oder steigendem cardiac output. In einem positiven Fall soll eine Therapie mit Kalziumkanalblocker initiiert, und die Messung nach 3-4 Monaten wiederholt werden. Bei Patienten mit anderen ätiologischen Gruppen ist diese Messung nicht empfohlen (16).
Im CT Thorax sind eine Dilatation des rechten Ventrikels und/oder des rechten Vorhofs, eine dilatierte Pulmonalarterie mit einem Durchmesser von mehr als 29 mm und ein erhöhter Quotient des Durchmessers von Pulmonalarterie/Aorta (>1) typisch für eine PH (17). Im CT Thorax können auch andere Lungenkrankheiten erkannt werden, wie Emphysem, Lungenfibrose, Gefässmalformationen, akute oder chronische Lungenembolie. Eine Ventilation-/Perfusion-Szintigraphie oder ein dual-energy CT wird empfohlen, um chronische Lungenembolie als Ursache der PH (CTEPH) zu suchen. Eine Lebersonographie wird bei Verdacht auf portale Hypertonie durchgeführt. Nach gesicherter Diagnosis kann ein kardiopulmonaler Belastungstest (Spiroergometrie) nützlich sein, um die Einschränkung der körperlicheren Belastbarkeit zu objektivieren und damit die Prognose vorherzusagen.
Therapie
Therapie der PAH (Gruppe 1): Nichtspezifische Therapie
Die Rechtsherzinsuffizienz ist assoziiert mit einer Hypervolämie, reduzierter renaler Durchblutung und der Aktivierung des Renin-Angiotensin-Aldosteron-Systems. Die Vermeidung und die Therapie der Wasserretention mittels diuretischer Therapie ist zentral bei PAH-Patienten (4). Die häufig eingesetzte Medikamentengruppe ist die Gruppe der Schleifendiuretika, welche gut mit anderen Diuretika, insbesondere Spironolactone kombiniert werden können (18). Die Verabreichungsform ist bei chronischer Rechtsherzbelastung oral (19) und muss eng überwacht werden mit täglicher Gewichtsmessung und regelmässigen Kontrollen der Elektrolyten und der Nierenfunktion. Bei Aszites und Ödem der gastrointestinalen Darmmukosa mit verminderter gastrointestinaler Absorption ist eine intravenöse Gabe von Lasix als Perfusor in der Akutphase angebracht (20).
Die meisten Patienten mit einer PH haben in Ruhe eine leicht reduzierte Blutoxygenierung, welche sich bei Belastung oder im Schlaf deutlich akzentuieren kann. Pathophysiologisch trägt diese Hypoxämie auch über eine verminderte gemischtvenöse Sauerstoffsättigung zusätzlich zur pulmonalen Vasokonstriktion bei und sollte daher wenn möglich mittels Sauerstoffgabe korrigiert werden (18). Randomisierte, kontrollierte Studien haben gezeigt, dass die Sauerstofftherapie bei pulmonal-vaskuläre Patienten die Belastbarkeit in der Fahrradergometrie signifikant verbessert und dass bei Patienten mit nächtlicher Hypoxämie (mittlere Sättigung < 92 %) und belastungsinduzierter Hypoxämie (Desaturation > 3 % < 92 %) eine Sauerstofftherapie über wenige Wochen die Belastbarkeit und Lebensqualität verbesserte (19) (21) (22). In den Guidelines wird die Sauerstofftherapie in Anlehnung an ältere Studien bei COPD-Patienten empfohlen, wenn bei einem PaO2 ≤ 8 kPa, respektive 60 mmHg oder bei einer SpO2 < 92 % (4) (23) (24). Im Falle einer Desaturation während der körperlichen Belastung oder wenn die Symptome dadurch gelindert werden können, kann eine ambulante Sauerstofftherapie in Erwägung gezogen werden.
Die Entscheidung über die Antikoagulationstherapie soll individuell, von Patient zu Patient getroffen werden, da keine robusten Daten vorliegen (25). Antikoaguliert werden sollen alle Patienten mit CTEPH und gewisse Patienten mit IPAH. Bei PAH mit systemischer Sklerose konnte sogar eine Mortalitätserhöhung den antikoagulierten Patienten nachgewiesen werden und ein negativer Effekt der Antikoagulation fand sich auch bei Patienten mit Lungenfibrose (26).
Bei Patienten mit pulmonal-vaskulären Krankheiten (PAH und CTEPH) wie bei fast allen PH-Formen konnte eine Verbesserung der Lebensqualität und der körperlichen Belastbarkeit durch eine sehr gezielte physische Aktivität und/oder Physiotherapieprogramme in Studien gezeigt werden (20). Eine moderate körperliche Aktivität ist daher im Rahmen der Symptome empfehlenswert, sofern sie nicht zur Dyspnoe oder thorakalen Schmerzen führen. Bei dekonditionierten Patienten kann eine überwachte Physiotherapie im stationären Setting empfohlen werden (27).
Aufgrund des chronischen Krankheitsverlaufs und erhöhten Gefahrensituation sollen alle Patienten gegen Influenza, Covid-19 und Pneumococcus geimpft werden (28). Jeden Patienten soll dringlich der Rauchstopp nahegelegt werden. Ein normaler Body Mass Index, sowie eine ausgewogene Diät sind wünschenswert. Viele Patienten brauchen psychologische Unterstützung, um eine Angststörung oder depressive Episode vermeiden zu können (27).
Therapie der PAH (Gruppe 1): Spezifische Therapie
Die spezifische PAH Therapie der PAH und CTEPH soll nur in spezialisierten Zentren eingeleitet werden, dies ist aufgrund der Komplexität der Diagnose und Therapie angebracht. Der Therapieverlauf soll zudem regelmässig mittels geeigneten Tests insbesondere Sechs-Minuten-Gehtest, Funktionsfragebögen, Echokardiografie, Spiroergometrie, ggf. Herz-MRI oder Wiederholung der Rechtsherzkatheteruntersuchung überwacht werden.
Die Vasoreaktivitätstestung wurde vorher schon erwähnt. Bei positivem Resultat werden die Patienten mittels einem Kalziumantagonist (Amlodipin, Nifedipin, Diltiazem, Felodipin) behandelt mit langsamer Auftitrierung bis zu einer höhen Dosis (z.B. Amlodipin Startdosis 5 mg, Zieldosis 15-30 mg/Tag). Eine Reevaluation der Therapieantwort ist nach 3-4 Monaten indiziert, bei fehlender Wirkung muss prompt auf eine spezifische PAH-Therapieoption umgestellt werden.
Bei negativer Vasoreaktivitätstestung oder bei Patienten ohne diesbezügliche Indikation werden PAH-spezifische Therapien eingesetzt.
Die folgenden Substanzen sind für die Therapie der PAH und zum Teil auch für die CTEPH zugelassen und wirken auf die drei pathophysiologischen Pathways: NO-sGC-cGMP Pathway: Phosphodiesterase-5-Hemmer (Sildenafil, Tadalafil, Vardenafil), und Stimulator der löslichen Guanylatcyclase (Riociguat), Endothelin-Pathway: Endothelin-Rezeptor-Antagonisten (Ambrisentan, Bosentan, Macitentan), Prostacyclin-Pathway: Prostacyclin-Analoga (Iloprost, Epoprostenol, Treprostinil) und Prostacyclin-Rezeptor-Agonisten (Selexipag) (29).
Der Algorithmus der zuletzt erschienenen Guidelines (ESC/ERS Guidelines 2022, (2)) unterscheidet Patienten ohne und mit kardiopulmonalen Komorbiditäten. Bei Patienten ohne kardiopulmonale Komorbiditäten wird eine Risikostratifizierung durchgeführt. Wenn der Patient sich in der tiefen oder intermediären Risikogruppe befindet, wird eine orale Doppeltherapie mit Endothelin-Rezeptor Antagonist und Phosphodiesterase-5-Hemmer begonnen. Bei Patienten in hoher Risikogruppe wird diese Therapie zusätzlich mit intravenösen oder subkutanen Prostazyklin-Analoga initiiert.
Regelmässige klinische Kontrollen, Reevaluation des klinischen Ansprechens und Assessment der Risikoklasse sind zentral und sollen alle 3-6 Monate stattfinden. Wenn die Patientin sich in der tiefen Risikoklasse befindet, kann die Therapie unverändert belassen werden. Wenn die Patientin in einer intermediären oder höheren Risikogruppe sich befindet, kann entweder ein Prostazyklin-Rezeptor-Agonist oder ein Switch von Phosphodiesterase-5-Hemmer zum Stimulator der löslichen Guanylatcyclase vorgenommen werden. Bei Patientinnen die sich klinisch verschlechtern, werden intravenöse oder subkutane Prostazyklin Analoga verabreicht.
Die intravenös verabreichten Prostanoide (häufig kommt Treprostinil zum Einsatz) werden durch einen zentral venösen Katheter kontinuierlich appliziert, mit der Hilfe einer Infusionspumpe (28) oder durch die Implantation einer subkutanen Lenus-Pro-Pumpe mit monatlichem Ausfüllen. Die Hauptnebenwirkungen sind Kieferschmerzen,
Diarrhoe, Flush und Gelenkschmerzen. Das subkutane Treprostinil ist eine mögliche Alternativtherapie.
Die Lungentransplantation bedeutet das ultima ratio für Patientinnen mit refraktärer PH. Aufgrund den erreichbaren, effektiven Therapiemöglichkeiten ist die Zahl an Lungentransplantationen mit dieser Indikation reduziert.
In der Risikostratifizierung finden sich klinische Kriterien (Zeichen einer Rechtsherzinsuffizienz, Progression den Symptomen, Synkope, NYHA-Klasse), Kriterien aus Belastungstesten (Gehstrecke während des Sechs-Minuten- Gehtestes, Leistungsfähigkeit in der Spiroergometrie), Laborwerte (BNP, NT-proBNP), echokardiographische Werte (rechtsventrikuläre Funktion, Perikarderguss), MRI-Messungen und hämodynamische Werte. Alle Kriterien wurden aufgrund ihrer prognostischen Werte selektioniert. Zahlreiche Studien haben gezeigt, dass sich die Prognose zwischen den verschiedenen Risikoklassen stark voneinander unterscheidet. Therapieziel muss es deshalb sein, die Patient*Innen in eine tiefere Risikoklasse zu bringen(2).
Die Komorbiditäten, die eine PH verschlechtern können, sollten ebenfalls behandelt werden. Die Begleitung und Betreuung durch spezialisierte Pflegefachpersonen ist bei dieser komplexen Therapien sehr hilfreich und von den Patient*Innen entsprechend positiv erlebt.
Neue Therapiemöglichkeiten
Die bereits erwähnten Mutationen im BMPR2 könnten zukünftig eine gezielte Therapie ermöglichen. Das Präparat Sotatercept soll in diesem Kontext das Gleichgewicht zwischen den wachstumsfördernden Aktivin Pathway und den wachstumshemmenden BMP Pathway wiederherstellen. Eine Phase-2-Studie hat bereits einen positiven, senkenden Effekt auf den pulmonalen vaskulären Widerstand gezeigt (30). Die aktuell laufende STELLAR-Studie (Phase 3) untersucht den Einfluss dieses Effekts auf die Belastungskapazität und Symptome (31).
Zurück zum Vignette-Fall
Die Patientin mit Sklerodermie wurde mittels Rechtsherzkatheter-Untersuchung weiter abgeklärt. Es zeigte sich eine prä-kapilläre PH: mPAP 32 mmHg, PAWP 8 mmHg, CO 3.8 l/min, CI 2.3 l/min/m2, PVR 6.3 WU. Das NT-pro-BNP war leicht erhöht mit 600 ng/L, klinisch mit einer Dyspnoe NYHA Grad II. Die ergänzenden Untersuchungen konnten eine Lungenfibrose, chronische Lungenembolie, HIV und Linksherzkrankheit ausschliessen. Die Patientin befindet sich in der tiefen bis intermediären Risikogruppe. Eine orale Doppeltherapie mit Tadalafil und Macitentan wurde sequentiell begonnen und gut vertragen. Darunter verbesserte sich die Leistungsfähigkeit (Dyspnoe NYHA I, Gehstrecke 500m) und das NT-pro-BNP normalisierte sich (<300ng/L). Damit befindet sich die Patientin in einer tiefen Risikoklasse, die Therapie wird unverändert weitergeführt und Kontrollen finden alle 6 Monaten statt.
Therapie der PH Gruppe 3
Die Therapie soll die Behandlung der zugrundeliegenden pulmonalen Erkrankung erzielen. Ergänzend ist die Indikation einer nicht invasiven oder invasiven Ventilation (CPAP/BiPAP) zu stellen. Je nach individuellem Entscheid kann eine pulmonale Rehabilitation angeboten werden.
Therapie der PH Gruppe 4
Patientinnen mit bewiesener CTEPH brauchen eine lebenslange Antikoagulation. Durch ein multimodales Team soll eine interdisziplinäre Entscheidung über die weitere Therapie erfolgen. Die Operabilität soll beurteilt werden. Falls eine Operation oder eine interventionelle Ballonangioplastie nicht in die Frage kommt, soll die konservative, medikamentöse Therapie unverzüglich eingeleitet werden. Im Falle einer Operationsindikation ist eine pulmonale Endarterektomie der nächste Schritt. Bei trotzdem persistierende PH und Symptomen infolge der konkomittierenden Kleingefässkrankheit bleibt nur die langjährige medikamentöse Therapie mit regelmässigen Nachkontrollen übrig.
Therapie der PH Gruppe 2
Die einzige Therapie ist die optimale, konsequente und multimodale Therapie der Linksherzinsuffizienz. Da die Hypervolämie ein wichtiges Merkmal ist und die Abnahme der rechtsventrikulären Funktion die Prognose der Herzinsuffizienz verschlechtert spielen die Diuretika eine zentrale Rolle. Die Einsetzung den klassischen PH-Medikamente ist nicht durch Evidenz untermauert, daher nicht indiziert und könnte für diese Patienten schädlich sein (2).
Prognose
Die Prognose der PH hängt von der jeweiligen Krankheitsgruppe ab, sie ist aber grundsätzlich eingeschränkt. Die Prognose kann mittels REVEAL score oder der oben beschriebenen Risikoklassifikation abgeschätzt werden (31). Nur
57% der Patienten leben nach 5 Jahren noch (2) (32). Die Mortalität in den USA hat seit 1980 von 5,2 auf 5,4 pro 100000 zugenommen (33).
Die Krankheitsgruppe 1 hat grundsätzlich eine schlechtere Überlebensrate als die anderen Gruppen. Innerhalb der PAH hat die idiopathische Form bessere Überlebenschancen, als die mit anderen Krankheiten assoziierten Formen. Das Eisenmenger Syndrom hat eine vergleichsweise bessere Prognose. Die Gruppe 2 hat ähnliche Prognosedaten wie Patient*Innen der Gruppe 1. Die Gruppe 3 (PH assoziiert mit chronische Lungenerkrankungen) hat eine deutlich reduzierte Prognose im Vergleich zur Gruppe 1 (38 versus 59% 5-Jahres-Überlebensrate). Die Gruppe 4 (chronisch thromboembolische pulmonale Hypertonie) weist die beste Überlebensraten auf, nach 5 Jahren leben 67% der Patienten (34).
Konklusion
Die PH ist eine chronische Erkrankung, die oft spät diagnostiziert wird und unbehandelt eine vergleichsweise hohe Mortalität aufweist. In der Differentialdiagnose der Dyspnoe soll immer an eine PH gedacht werden. Die frühe Abklärung ermöglicht die Verdachtsdiagnose mittels TTE und Verifizierung in der Rechtsherzkatheteruntersuchung. Besonders gefährdete Krankheitsgruppen (z.B. Patient*Innen mit Sklerodermie) sollten regelmässig gescreent werden. Dank breiten Behandlungsoptionen sind die Überlebenschancen unter Therapie deutlich besser. Aufgrund der Komplexität der Erkrankung ist die Behandlung der PAH und der CTPEH in spezialisierten Zentren durchzuführen.
Klinik Innere Medizin und Pneumologie, Stadtspital Zürich Waid
Tièchestrasse 99
8037 Zürich
Charlotte.Berlier@stadtspital.ch
1. Kroegel C, Costabel U. Klinische Pneumologie. Das Referenzwerk für Klinik und Praxis. 1st ed.2014;5950.
2 Humbert M, Kovacs G, Hoeper M, Badagliacca R, Berger R, Brida M. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. European Heart Journal. 2022;00:1–114.
3 Huber L, Vrugt B, Arrigo M. Pulmonary hypertension: Classification and pathobiology. Cardiovascular Medicine. 2014;17(11):312–319.
4 Prella M, Yerlyb P, Auberta J. Treatment of pulmonary arterial hypertension: an update. Cardiovascular Medicine. 2014;17(11):334–343.
5 Neumann M, Wong K, Lazo K, Stover D. Iron therapy as a novel treatment of scleroderma-related pulmonary hypertension: A case report and literature review. Respiratory case reports. 2022;10:e0904.
6 Schwiening M, Swietlik E, Pandya D, Burling K, Barker P, Feng O, et al. Different cytokine patterns in BMPR2-mutation positive patients and pulmonary arterial hypertension patients without mutations and their influence on survival. CHEST. 2022; 161(6):1651-1656.
7 Melzig C, Do T D, Egenlauf B, Fink M, Grünig E, Kauczor H, et al. Utility of automated cardiac chamber volumetry by non-gated CT pulmonary angiography for detection of pulmonary hypertension using the 2018 Updated Hemodynamic Definition. American Journal of Roentgology. 2022;219:66-75.
8 Klabunde R. Normal and abnormal blood pressure (Physiology, Pathophysiology & Treatment). 1st ed. 2013.
9 Kovacs G, Herve P, Barbera J, Chaouat A, Chemla D, Condliffe R et al. An official European Respiratory Society statement: pulmonary haemodynamics during exercise. Eur Respir J 2017,50: 1700578.
10 Apitz C, Kozlik-Feldmann R, Kaemmerer H, Gorenflo M, Lammers A. Pulmonale Hypertonie. S2k Leitlinie der deutschen Gesellschaft für pädiatrische Kardiologie und angeborene Herzfehler. AWMF online. 2020;34.
11 Ulrich S, Hersberger M, Fischler M, Nussbaumer-Ochsner Y, Treder U, Russi E et al. Genetic polymorphisms of the serotonin transporter, but not the 2a receptor or nitric oxide synthetase, are associated with pulmonary hypertension in chronic obstructive pulmonary disease. Respiration. 2010;79(4):288-95.
12 Humbert M, Guignabert C, Bonnet S, Dorfmüller P, Klinger J, Nicolls M, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. European Respiratory Journal. 2019;53:1801887.
13 Eddahibi S, Chaouat A, Morrell N, Fadel E, Fuhrman C, Bugnet A, et al. Polymorphism of the serotonin transporter gene and pulmonary hypertension in chronic obstructive pulmonary disease. Circulation. 2003;108:1839-1844.
14 Olschewski A, Berghausen E.M, Eichstaedt C.A, Fleischmann B.K, Grünig E, Grünig G, et al. Pathobiologie, Pathologie und Genetik der pulmonalen Hypertonie. Dtsch Med Wochenschr. 2016;141:4-9.
15 American College of Chest Physicians. ACCP Pulmonary Medicine Board Review: 26th ed. 2012. 41.
16 Frost A, Badesch D, Gibbs J, Gopalan D, Khanna D, Manes A, et al. Diagnosis of pulmonary hypertension. Eur Respir J. 2019;53:1801904.
17 Noordegraaf A, Chin K, Haddad F, Hassoun P, Hemnes A, Hopkins S, et al. Pathophysiology of the right ventricle and of the pulmonary circulation in pulmonary hypertension: an update. Eur Respir J. 2019;53:1801900.
18 Carta A, Lichtblau M, Berlier C, Saxer S, Schneider S, Schwarz E et al. The Impact of Breathing Hypoxic Gas and Oxygen on Pulmonary Hemodynamics in Patients With Pulmonary Hypertension. Front Med (Lausanne). 2022, 9: 791423.
19 Ulrich S, Saxer S, Hasler E, Schwarz E, Schneider S, Furian M et al. Effect of domiciliary oxygen therapy on exercise capacity and quality of life in patients with pulmonary arterial or chronic thromboembolic pulmonary hypertension: a randomised, placebo-controlled trial. Eur Respir J. 2019,1;54(2):1900276.
20 Grünig E, Benjamin N, Krüger U, Kaemmerer H, Harutyunova S, Olsson K.M, et al. Allgemeine und supportive Therapie der pulmonal artriellen Hypertonie. Dtsch Med Wochenschr 2016;141:26-32.
21 Ulrich S, Hasler E, Saxer S, Furian M, Müller-Mottet S, Keusch S et al. Effect of breathing oxygen-enriched air on exercise performance in patients with precapillary pulmonary hypertension: randomized, sham-controlled cross-over trial. Eur Heart J. 2017,14;38(15):1159-1168.
22 Ulrich S, Keusch S, Hildenbrand F, Lo Cascio C, Huber L, Tanner F et al. Effect of nocturnal oxygen and acetazolamide on exercise performance in patients with pre-capillary pulmonary hypertension and sleep-disturbed breathing: randomized, double-blind, cross-over trial. Eur Heart J. 2015,7;36(10):615-23.
23 Rosenkranz S, Preston I. Right heart catheterisation: best practice und pitfalls in pulmonary hypertension. Eur Respir Rev. 2015;24:642-65.
24 Mereles D, Ehlken N, Kreuscher S, Ghofrani S, Hoeper M, Halank M, et al. Exercise and respiratory training improve exercise capacity and quality of life in patients with severe chronic pulmonary hypertension. Circulation. 2006;114:1482-1489.
25 Khan M, Usman M, Siddiqi T, Khan S, Murad M, Mookadam F, et al. Is antikoagulation beneficial in pulmonary arterial hypertension? Circ Cardiovasc Qual Outcomes. 2018;11(9):004757.
26 Hopkins W, Rubin L. Treatment and prognosis of pulmonary arterial hypertension in adults (group 1). (Internet). (abgerufen am 07.July 2022). Verfügbar unter: https://www.uptodate.com/contents/treatment-and-prognosis-of-pulmonary-arterial-hypertension-in-adults-group-1#H3048703378.
27 Gabriel L, Delavenne X, Bedouch P, Khouatra C, Bouvaist H, Cordier JF, et al. Risk of Direct Oral Anticoagulant Bioaccumulation in Patients with Pulmonary Hypertension. Respiration. 2016;91(4):307-15.
28 Brast RJ, Gibbs JS, Ghofrani HA, Hoeper MM; McLaughlin VV, Rubin LJ, et al. Updated evidence-based treatment algorithm in pulmonary arterial hypertension. J. Am Coll Cardiol. 2009;54:78-84.
29 Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, McGoon MD. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest. 2012;142(2):448.
30 Humbert M, McLaughlin V, Gibbs S, Gomberg-Maitland M, Hoeper M, Preston I et al. Sotatercept for the treatment of pulmonary arterial hypertension. New England Journal of Medicine. 2021, 384;13.
31 Hopkins W, Rubin L. Treatment of pulmonary arterial hypertension (group 1) in adults: Pulmonary-hypertension specific therapy. (Internet). (abgerufen am 19.August 2022). Verfügbar unter: https://www.uptodate.com/contents/treatment-of-pulmonary-arterial-hypertenion-group-1-in-adults-
pulmonary-hypertension-specific-therapy?topicRef=121912&source=see_link#H2820057861.
32 Hoeper M, Benza R, Corris P, de Perrot M, Fadel E, Keogh A. et al. Intensive care, right ventricular support and lung transplantation in patients with pulmonary hypertension. Eur Respir J. 2019;53:1801906.
33 Hyduk A, Croft JB, Ayala C, Zheng K, Zheng ZJ, Mensah GA, et al. Pulmonary hypertension surveillance–United States, 1980-2002. MMWR Surveill Summ. 2005;54(5):1.
34 Gall H, Felix JF, Schneck FK, Milger K, Sommer N, Voswinckel R, et al.
The Giessen Pulmonary Hypertension Registry: Survival in pulmonary hypertension subgroups. J Heart Lung Transplant. 2017;36(9):957.